 +
+Each row is a gene that is a member of module 19, and the composite expression of these genes, called an eigengene, is shown in the barplot below.
+This plot demonstrates how these genes, together as a module, are differentially expressed between our two time points.
+
+**IDENTICAL TO MICROARRAY EXAMPLE**
+## Check out our file structure!
+
+Your new analysis folder should contain:
+
+- The example analysis `.Rmd` you downloaded
+- A folder called `data` (currently empty)
+- A folder for `plots` (currently empty)
+- A folder for `results` (currently empty)
+
+Your example analysis folder should contain your `.Rmd` and three empty folders (which won't be empty for long!).
+
+If the concept of a "file path" is unfamiliar to you; we recommend taking a look at our [section about file paths](https://alexslemonade.github.io/refinebio-examples/01-getting-started/getting-started.html#an-important-note-about-file-paths-and-Rmds).
+
+**REVIEW**
+# Using a different refine.bio dataset with this analysis?
+
+If you'd like to adapt an example analysis to use a different dataset from [refine.bio](https://www.refine.bio/), we recommend replacing the `gene_module_url` with a different file path to a gene list that you are interested in.
+The file we use here has two columns from our WGCNA module results, so if your gene list differs, many steps will need to be changed or deleted entirely depending on what your gene list file looks like (particularly in the [`Determine our genes of interest list` section](#determined-our-genes-of-interest-list)).
+
+**IDENTICAL TO MICROARRAY EXAMPLE**
+We suggest saving plots and results to `plots/` and `results/` directories, respectively, as these are automatically created by the notebook.
+From here you can customize this analysis example to fit your own scientific questions and preferences.
+
+***
+
+
+
+Each row is a gene that is a member of module 19, and the composite expression of these genes, called an eigengene, is shown in the barplot below.
+This plot demonstrates how these genes, together as a module, are differentially expressed between our two time points.
+
+**IDENTICAL TO MICROARRAY EXAMPLE**
+## Check out our file structure!
+
+Your new analysis folder should contain:
+
+- The example analysis `.Rmd` you downloaded
+- A folder called `data` (currently empty)
+- A folder for `plots` (currently empty)
+- A folder for `results` (currently empty)
+
+Your example analysis folder should contain your `.Rmd` and three empty folders (which won't be empty for long!).
+
+If the concept of a "file path" is unfamiliar to you; we recommend taking a look at our [section about file paths](https://alexslemonade.github.io/refinebio-examples/01-getting-started/getting-started.html#an-important-note-about-file-paths-and-Rmds).
+
+**REVIEW**
+# Using a different refine.bio dataset with this analysis?
+
+If you'd like to adapt an example analysis to use a different dataset from [refine.bio](https://www.refine.bio/), we recommend replacing the `gene_module_url` with a different file path to a gene list that you are interested in.
+The file we use here has two columns from our WGCNA module results, so if your gene list differs, many steps will need to be changed or deleted entirely depending on what your gene list file looks like (particularly in the [`Determine our genes of interest list` section](#determined-our-genes-of-interest-list)).
+
+**IDENTICAL TO MICROARRAY EXAMPLE**
+We suggest saving plots and results to `plots/` and `results/` directories, respectively, as these are automatically created by the notebook.
+From here you can customize this analysis example to fit your own scientific questions and preferences.
+
+***
+
+ Over-representation analysis - RNA-Seq
+CCDL for ALSF
+December 2020
+ +IDENTICAL TO MICROARRAY EXAMPLE (except microarray –> rnaseq) # Purpose of this analysis
+This example is one of pathway analysis module set, we recommend looking at the pathway analysis table below to help you determine which pathway analysis method is best suited for your purposes.
+This particular example analysis shows how you can use over-representation analysis (ORA) to determine if a set of genes shares more or fewer genes with gene sets/pathways than we would expect by chance.
+ORA is a broadly applicable technique that may be good in scenarios where your dataset or scientific questions don’t fit the requirements of other pathway analyses methods. It also does not require any particular sample size, since the only input from your dataset is a set of genes of interest (Yaari et al. 2013).
+If you have differential expression results or something with a gene-level ranking and a two-group comparison, we recommend considering GSEA for your pathway analysis questions. ⬇️ Jump to the analysis code ⬇️
+0.0.1 What is pathway analysis?
+Pathway analysis refers to any one of many techniques that uses predetermined sets of genes that are related or coordinated in their expression in some way (e.g., participate in the same molecular process, are regulated by the same transcription factor) to interpret a high-throughput experiment. In the context of refine.bio, we use these techniques to analyze and interpret genome-wide gene expression experiments. The rationale for performing pathway analysis is that looking at the pathway-level may be more biologically meaningful than considering individual genes, especially if a large number of genes are differentially expressed between conditions of interest. In addition, many relatively small changes in the expression values of genes in the same pathway could lead to a phenotypic outcome and these small changes may go undetected in differential gene expression analysis.
+We highly recommend taking a look at Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges from Khatri et al. (2012) for a more comprehensive overview. We have provided primary publications and documentation of the methods we will introduce below as well as some recommended reading in the Resources for further learning section.
0.0.2 How to choose a pathway analysis?
+This table summarizes the pathway analyses examples in this module.
+| Analysis | +What is required for input | +What output looks like | +✅ Pros | +⚠️ Cons | +
|---|---|---|---|---|
| ORA (Over-representation Analysis) | +A list of gene IDs (no stats needed) | +A per-pathway hypergeometric test result | +- Simple - Inexpensive computationally to calculate p-values |
+- Requires arbitrary thresholds and ignores any statistics associated with a gene - Assumes independence of genes and pathways |
+
| GSEA (Gene Set Enrichment Analysis) | +A list of genes IDs with gene-level summary statistics | +A per-pathway enrichment score | +- Includes all genes (no arbitrary threshold!) - Attempts to measure coordination of genes |
+- Permutations can be expensive - Does not account for pathway overlap - Two-group comparisons not always appropriate/feasible |
+
| GSVA (Gene Set Variation Analysis) | +A gene expression matrix (like what you get from refine.bio directly) | +Pathway-level scores on a per-sample basis | +- Does not require two groups to compare upfront - Normally distributed scores |
+- Scores are not a good fit for gene sets that contain genes that go up AND down - Method doesn’t assign statistical significance itself - Recommended sample size n > 10 |
+
1 How to run this example
+For general information about our tutorials and the basic software packages you will need, please see our ‘Getting Started’ section. We recommend taking a look at our Resources for Learning R if you have not written code in R before.
+1.1 Obtain the .Rmd file
+To run this example yourself, download the .Rmd for this analysis by clicking this link.
Clicking this link will most likely send this to your downloads folder on your computer. Move this .Rmd file to where you would like this example and its files to be stored.
You can open this .Rmd file in RStudio and follow the rest of these steps from there. (See our section about getting started with R notebooks if you are unfamiliar with .Rmd files.)
1.2 Set up your analysis folders
+Good file organization is helpful for keeping your data analysis project on track! We have set up some code that will automatically set up a folder structure for you. Run this next chunk to set up your folders!
+If you have trouble running this chunk, see our introduction to using .Rmds for more resources and explanations.
# Create the data folder if it doesn't exist
+if (!dir.exists("data")) {
+ dir.create("data")
+}
+
+# Define the file path to the plots directory
+plots_dir <- "plots" # Can replace with path to desired output plots directory
+
+# Create the plots folder if it doesn't exist
+if (!dir.exists(plots_dir)) {
+ dir.create(plots_dir)
+}
+
+# Define the file path to the results directory
+results_dir <- "results" # Can replace with path to desired output results directory
+
+# Create the results folder if it doesn't exist
+if (!dir.exists(results_dir)) {
+ dir.create(results_dir)
+}In the same place you put this .Rmd file, you should now have three new empty folders called data, plots, and results!
REVIEW
+1.3 About the dataset we are using for this example
+For this example analysis, we will use this acute viral bronchiolitis dataset. The data that we downloaded from refine.bio for this analysis has 62 paired peripheral blood mononuclear cell RNA-seq samples obtained from 31 patients. Samples were collected at two time points: during their first, acute bronchiolitis visit (abbreviated “AV”) and their recovery, their post-convalescence visit (abbreviated “CV”).
+1.4 Obtain the gene set for this example
+In this example, we are using a set of co-expressed genes we obtained from an example analysis of acute viral bronchiolitis dataset using WGCNA (Langfelder and Horvath 2008).
We have provided this file for you and the code in this notebook will read in the results that are stored online, but if you’d like to follow the steps for obtaining this results file yourself, we suggest going through that WGCNA example.
+In this example, we will focus on one gene module, called module 19, to see what pathways these genes in this module may be associated with. We identified this module 19 as the most differentially expressed between our data’s two time points (during illness and recovering from illness).
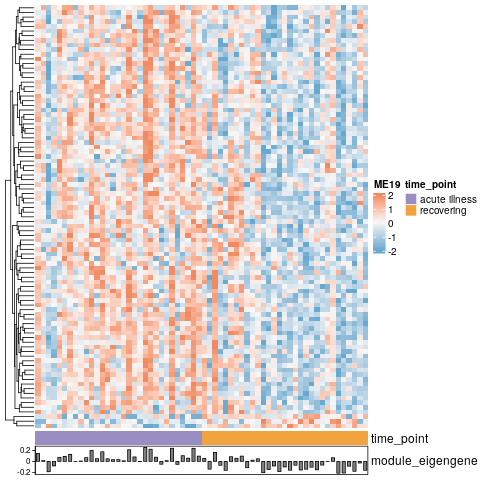
+Here’s a heatmap to show a preview of this module and the genes that make it up.
+
Each row is a gene that is a member of module 19, and the composite expression of these genes, called an eigengene, is shown in the barplot below. This plot demonstrates how these genes, together as a module, are differentially expressed between our two time points.
+IDENTICAL TO MICROARRAY EXAMPLE ## Check out our file structure!
+Your new analysis folder should contain:
+-
+
- The example analysis
.Rmdyou downloaded
+
+ - A folder called
data(currently empty)
+ - A folder for
plots(currently empty)
+
+ - A folder for
results(currently empty)
+
Your example analysis folder should contain your .Rmd and three empty folders (which won’t be empty for long!).
If the concept of a “file path” is unfamiliar to you; we recommend taking a look at our section about file paths.
+REVIEW # Using a different refine.bio dataset with this analysis?
+If you’d like to adapt an example analysis to use a different dataset from refine.bio, we recommend replacing the gene_module_url with a different file path to a gene list that you are interested in. The file we use here has two columns from our WGCNA module results, so if your gene list differs, many steps will need to be changed or deleted entirely depending on what your gene list file looks like (particularly in the Determine our genes of interest list section).
IDENTICAL TO MICROARRAY EXAMPLE We suggest saving plots and results to plots/ and results/ directories, respectively, as these are automatically created by the notebook. From here you can customize this analysis example to fit your own scientific questions and preferences.
+ + +
2 Over-Representation Analysis with clusterProfiler - RNA-seq
+2.1 Install libraries
+See our Getting Started page with instructions for package installation for a list of the other software you will need, as well as more tips and resources.
+In this analysis, we will be using clusterProfiler package to perform ORA and the msigdbr package which contains gene sets from the Molecular Signatures Database (MSigDB) already in the tidy format required by clusterProfiler (Dolgalev 2020; Subramanian et al. 2005).
We will also need the org.Hs.eg.db package to perform gene identifier conversion and ggupset to make an UpSet plot (Carlson 2019; Ahlmann-Eltze 2020).
if (!("clusterProfiler" %in% installed.packages())) {
+ # Install this package if it isn't installed yet
+ BiocManager::install("clusterProfiler", update = FALSE)
+}
+
+# This is required to make one of the plots that clusterProfiler will make
+if (!("ggupset" %in% installed.packages())) {
+ # Install this package if it isn't installed yet
+ BiocManager::install("ggupset", update = FALSE)
+}
+
+if (!("msigdbr" %in% installed.packages())) {
+ # Install this package if it isn't installed yet
+ BiocManager::install("msigdbr", update = FALSE)
+}
+
+if (!("org.Hs.eg.db" %in% installed.packages())) {
+ # Install this package if it isn't installed yet
+ BiocManager::install("org.Hs.eg.db", update = FALSE)
+}Attach the packages we need for this analysis.
+ +## ## clusterProfiler v3.18.0 For help: https://guangchuangyu.github.io/software/clusterProfiler
+##
+## If you use clusterProfiler in published research, please cite:
+## Guangchuang Yu, Li-Gen Wang, Yanyan Han, Qing-Yu He. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS: A Journal of Integrative Biology. 2012, 16(5):284-287.##
+## Attaching package: 'clusterProfiler'## The following object is masked from 'package:stats':
+##
+## filter# Package that contains MSigDB gene sets in tidy format
+library(msigdbr)
+
+# Homo sapiens annotation package we'll use for gene identifier conversion
+library(org.Hs.eg.db)## Loading required package: AnnotationDbi## Loading required package: stats4## Loading required package: BiocGenerics## Loading required package: parallel##
+## Attaching package: 'BiocGenerics'## The following objects are masked from 'package:parallel':
+##
+## clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
+## clusterExport, clusterMap, parApply, parCapply, parLapply,
+## parLapplyLB, parRapply, parSapply, parSapplyLB## The following objects are masked from 'package:stats':
+##
+## IQR, mad, sd, var, xtabs## The following objects are masked from 'package:base':
+##
+## anyDuplicated, append, as.data.frame, basename, cbind, colnames,
+## dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
+## grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget,
+## order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
+## rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply,
+## union, unique, unsplit, which.max, which.min## Loading required package: Biobase## Welcome to Bioconductor
+##
+## Vignettes contain introductory material; view with
+## 'browseVignettes()'. To cite Bioconductor, see
+## 'citation("Biobase")', and for packages 'citation("pkgname")'.## Loading required package: IRanges## Loading required package: S4Vectors##
+## Attaching package: 'S4Vectors'## The following object is masked from 'package:clusterProfiler':
+##
+## rename## The following object is masked from 'package:base':
+##
+## expand.grid##
+## Attaching package: 'IRanges'## The following object is masked from 'package:clusterProfiler':
+##
+## slice##
+## Attaching package: 'AnnotationDbi'## The following object is masked from 'package:clusterProfiler':
+##
+## select## REVIEW ## Import data
+For ORA, we only need a list of gene IDs as our input, so this example can work for any situations where you have gene list and want to know more about what biological pathways it shares genes with.
+For this example, we will read in results from a co-expression network analysis that we have already performed. Rather than reading from a local file, we will download the results table directly from a URL. These results are from a acute bronchiolitis experiment we used for an example WGCNA analysis (Langfelder and Horvath 2008). The table contains two columns, one with Ensembl gene IDs, and the other with the name of the module they are a part of. We will perform ORA on one of the modules we identified in the WGCNA analysis. In order to know which genes are a part of this module, we will import a saved results file that’s stored online and contains the genes and what module they were identified to be a part of.
+Instead of using this URL below, you can use a file path to a TSV file with your desired gene list. First we will assign the URL to its own variable called, gene_module_url.
# Define the url to your gene list file
+gene_module_url <- "https://refinebio-examples.s3.us-east-2.amazonaws.com/04-advanced-topics/results/SRP140558_wgcna_gene_to_module.tsv"Read in the file that has WGCNA gene and module results. Here we are using the URL we set up above, but this can be a local file path instead i.e. you can replace gene_module_url in the code below with a path to file you have on your computer like: file.path("results", "SRP140558_results_gene_list.tsv").
# Read in the contents of your differential expression results file
+# `gene_module_url` can be replaced with a file path to a TSV file with your
+# desired gene list results
+gene_module_df <- readr::read_tsv(gene_module_url)##
+## ── Column specification ──────────────────────────────────────────────────────────────────────────────────────────────
+## cols(
+## gene = col_character(),
+## module = col_character()
+## )read_tsv() can read TSV files online and doesn’t necessarily require you download the file first. Let’s take a look at what this gene module list looks like.
IDENTICAL TO MICROARRAY EXAMPLE (except Dr –> Hs) ## Getting familiar with clusterProfiler’s options
Let’s take a look at what organisms the package supports.
+ +The data we’re interested in here comes from human samples, so we can obtain just the gene sets relevant to H. sapiens with the species argument to msigdbr().
MSigDB contains 8 different gene set collections (Subramanian et al. 2005).
+H: hallmark gene sets
+C1: positional gene sets
+C2: curated gene sets
+C3: motif gene sets
+C4: computational gene sets
+C5: GO gene sets
+C6: oncogenic signatures
+C7: immunologic signaturesIn this example, we will use pathways that are gene sets considered to be “canonical representations of a biological process compiled by domain experts” and are a subset of C2: curated gene sets (Subramanian et al. 2005).
Specifically, we will use the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways (Kanehisa and Goto 2000).
+First, let’s take a look at what information is included in this data frame.
+ +We will need to use gs_cat and gs_subcat columns to construct a filter step that will only keep curated gene sets and KEGG pathways.
# Filter the human data frame to the KEGG pathways that are included in the
+# curated gene sets
+hs_kegg_df <- hs_msigdb_df %>%
+ dplyr::filter(
+ gs_cat == "C2", # This is to filter only to the C2 curated gene sets
+ gs_subcat == "CP:KEGG" # This is because we only want KEGG pathways
+ )The clusterProfiler() function we will use requires a data frame with two columns, where one column contains the term identifier or name and one column contains gene identifiers that match our gene lists we want to check for enrichment.
Our data frame with KEGG terms contains Entrez IDs and gene symbols.
+In our differential expression results data frame, gene_module_df we have Ensembl gene identifiers. So we will need to convert our Ensembl IDs into either gene symbols or Entrez IDs.
2.2 Gene identifier conversion
+We’re going to convert our identifiers in gene_module_df to gene symbols because they are a bit more human readable, but you can, with the change of a single argument, use the same code to convert to many other types of identifiers!
The annotation package org.Hs.eg.db contains information for different identifiers (Carlson 2019). org.Hs.eg.db is specific to Homo sapiens – this is what the s` in the package name is referencing.
Take a look at our other gene identifier conversion examples for examples with different species and gene ID types: the microarray example and the RNA-seq example.
+We can see what types of IDs are available to us in an annotation package with keytypes().
## [1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT" "ENSEMBLTRANS"
+## [6] "ENTREZID" "ENZYME" "EVIDENCE" "EVIDENCEALL" "GENENAME"
+## [11] "GO" "GOALL" "IPI" "MAP" "OMIM"
+## [16] "ONTOLOGY" "ONTOLOGYALL" "PATH" "PFAM" "PMID"
+## [21] "PROSITE" "REFSEQ" "SYMBOL" "UCSCKG" "UNIGENE"
+## [26] "UNIPROT"Even though we’ll use this package to convert from Ensembl gene IDs (ENSEMBL) to Entrez IDs (ENTREZID), we could just as easily use it to convert from an Ensembl transcript ID (ENSEMBLTRANS) to gene symbols (SYMBOL).
The function we will use to map from Ensembl gene IDs to Entrez IDs is called mapIds().
# This returns a named vector which we can convert to a data frame, where
+# the keys (Ensembl IDs) are the names
+entrez_vector <- mapIds(org.Hs.eg.db, # Specify the annotation package
+ # The vector of gene identifiers we want to map
+ keys = gene_module_df$gene,
+ # The type of gene identifier we want returned
+ column = "ENTREZID",
+ # What type of gene identifiers we're starting with
+ keytype = "ENSEMBL",
+ # In the case of 1:many mappings, return the
+ # first one. This is default behavior!
+ multiVals = "first"
+)## 'select()' returned 1:many mapping between keys and columnsThis message is letting us know that sometimes Ensembl gene identifiers will map to multiple Entrez IDs. In this case, it’s also possible that an Entrez ID will map to multiple Ensembl IDs. For more about how to explore this, take a look at our RNA-seq gene ID conversion example.
+Let’s create a two column data frame that shows the gene symbols and their Ensembl IDs side-by-side.
+# We would like a data frame we can join to the differential expression stats
+gene_key_df <- data.frame(
+ ensembl_id = names(entrez_vector),
+ entrez_id = entrez_vector,
+ stringsAsFactors = FALSE
+) %>%
+ # If an Ensembl gene identifier doesn't map to a gene symbol, drop that
+ # from the data frame
+ dplyr::filter(!is.na(entrez_id))Let’s see a preview of entrez_id.
Now we are ready to add the gene_key_df to our data frame with the module labels, gene_module_df. Here we’re using a dplyr::left_join() because we only want to retain the genes that have Entrez IDs and this will filter out anything in our gene_module_df that does not have an Entrez ID when we join using the Ensembl gene identifiers.
module_annot_df <- gene_key_df %>%
+ # Using a left join removes the rows without gene symbols because those rows
+ # have already been removed in `gene_key_df`
+ dplyr::left_join(gene_module_df,
+ # The name of the column that contains the Ensembl gene IDs
+ # in the left data frame and right data frame
+ by = c("ensembl_id" = "gene")
+ )Let’s take a look at what this data frame looks like.
+ +2.3 Over-representation Analysis (ORA)
+Over-representation testing using clusterProfiler is based on a hypergeometric test (often referred to as Fisher’s exact test) (Yu). For more background on hypergeometric tests, this handy tutorial explains more about how hypergeometric tests work (Puthier and van Helden 2015).
We will need to provide to clusterProfiler two genes lists:
-
+
- Our genes of interest +
- What genes were in our total background set. (All genes that originally had an opportunity to be measured). +
2.4 Determine our genes of interest list
+REVIEW
+This step is highly variable depending on what your gene list is, what information it contains and what your goals are. You may want to delete this next chunk entirely if you supply an already determined list of genes OR you may need to adjust the cutoffs and column names.
+ORA generally requires you make some sort of arbitrary decision to obtain your genes of interest list and this is one of the approach’s weaknesses – to get to a gene list we’ve removed all other context.
+In this example, we will focus on one module, module 19, which we previously identified as differentially expressed between our time points (during the acute bronchiolitis illness and during recovery from bronchiolitis). See the previous section for more background on this gene list.
+module_19_genes <- module_annot_df %>%
+ dplyr::filter(module == "ME19") %>%
+ dplyr::pull("entrez_id")Because one entrez_id may map to multiple Ensembl IDs, we need to make sure we have no repeated Entrez IDs in this list.
# Reduce to only unique Entrez IDs
+genes_of_interest <- unique(as.character(module_19_genes))
+
+# Let's print out some of these genes
+head(genes_of_interest)## [1] "5704" "578" "23471" "5255" "4171" "8898"2.5 Determine our background set gene list
+Sometimes folks consider genes from the entire genome to comprise the background, but for this RNA-seq example, we will consider all detectable genes as our background set. The dataset that these genes are from already had unreliably detected, low count genes removed it. Because of this, we can obtain our detected genes list from our data frame, module_annot_df (which we have not done any further filtering on in this notebook).
IDENTICAL TO MICROARRAY EXAMPLE (except plot names changed and results summaries altered)
+2.6 Run ORA using the enricher() function
+Now that we have our background set, our genes of interest, and our pathway information, we’re ready to run ORA using the enricher() function.
kegg_ora_results <- enricher(
+ gene = genes_of_interest, # A vector of your genes of interest
+ pvalueCutoff = 0.1, # Can choose a FDR cutoff
+ pAdjustMethod = "BH", # What method for multiple testing correction should we use
+ universe = background_set, # A vector containing your background set genes
+ # The pathway information should be a data frame with a term name or
+ # identifier and the gene identifiers
+ TERM2GENE = dplyr::select(
+ hs_kegg_df,
+ gs_name,
+ human_entrez_gene
+ )
+)Note: using enrichKEGG() is a shortcut for doing ORA using KEGG, but the approach we covered here can be used with any gene sets you’d like!
The information we’re most likely interested in is in the results slot. Let’s convert this into a data frame that we can write to file.
Let’s print out a sneak peek of it here and take a look at how many sets do we have that fit our cutoff of 0.1 FDR?
Looks like there are four KEGG sets returned as significant at FDR of 0.1.
2.7 Visualizing results
+We can use a dot plot to visualize our significant enrichment results. The enrichplot::dotplot() function will only plot gene sets that are significant according to the multiple testing corrected p values (in the p.adjust column) and the pvalueCutoff you provided in the enricher() step.
## wrong orderBy parameter; set to default `orderBy = "x"`
Use ?enrichplot::dotplot to see the help page for more about how to use this function.
This plot is arguably more useful when we have a large number of significant pathways.
+Let’s save it to a PNG.
+ggplot2::ggsave(file.path(plots_dir, "SRP140558_ora_enrich_plot_module_19.png"),
+ plot = enrich_plot
+)## Saving 7 x 5 in imageWe can use an UpSet plot to visualize the overlap between the gene sets that were returned as significant.
+ +
See that KEGG_CELL_CYCLE and KEGG_OOCYTE_MEIOSIS have genes in common, as do KEGG_CELL_CYCLE and KEGG_DNA_REPLICATION. Gene sets or pathways aren’t independent! Based on the context of your samples, you may be able to narrow down which ones make sense, like in this instance, we are dealing with PBMCs, so the oocyte meiosis is not relevant to the biology of the samples at hand.
Let’s also save this to a PNG.
+ggplot2::ggsave(file.path(plots_dir, "SRP140558_ora_upset_plot_module_19.png"),
+ plot = upset_plot
+)## Saving 7 x 5 in image3 Resources for further learning
+-
+
- Hypergeometric test exercises(Puthier and van Helden 2015). +
- clusterProfiler ORA tutorial +
- clusterProfiler paper (Yu et al. 2012). +
- clusterProfiler book (Yu). +
- This handy review which summarizes the different types of pathway analysis and their limitations (Khatri et al. 2012). +
4 Session info
+At the end of every analysis, before saving your notebook, we recommend printing out your session info. This helps make your code more reproducible by recording what versions of software and packages you used to run this.
+ +## ─ Session info ───────────────────────────────────────────────────────────────
+## setting value
+## version R version 4.0.2 (2020-06-22)
+## os Ubuntu 20.04 LTS
+## system x86_64, linux-gnu
+## ui X11
+## language (EN)
+## collate en_US.UTF-8
+## ctype en_US.UTF-8
+## tz Etc/UTC
+## date 2020-12-01
+##
+## ─ Packages ───────────────────────────────────────────────────────────────────
+## package * version date lib source
+## AnnotationDbi * 1.52.0 2020-10-27 [1] Bioconductor
+## assertthat 0.2.1 2019-03-21 [1] RSPM (R 4.0.0)
+## backports 1.1.10 2020-09-15 [1] RSPM (R 4.0.2)
+## Biobase * 2.50.0 2020-10-27 [1] Bioconductor
+## BiocGenerics * 0.36.0 2020-10-27 [1] Bioconductor
+## BiocManager 1.30.10 2019-11-16 [1] RSPM (R 4.0.0)
+## BiocParallel 1.24.1 2020-11-06 [1] Bioconductor
+## bit 4.0.4 2020-08-04 [1] RSPM (R 4.0.2)
+## bit64 4.0.5 2020-08-30 [1] RSPM (R 4.0.2)
+## blob 1.2.1 2020-01-20 [1] RSPM (R 4.0.0)
+## cli 2.1.0 2020-10-12 [1] RSPM (R 4.0.2)
+## clusterProfiler * 3.18.0 2020-10-27 [1] Bioconductor
+## colorspace 1.4-1 2019-03-18 [1] RSPM (R 4.0.0)
+## cowplot 1.1.0 2020-09-08 [1] RSPM (R 4.0.2)
+## crayon 1.3.4 2017-09-16 [1] RSPM (R 4.0.0)
+## curl 4.3 2019-12-02 [1] RSPM (R 4.0.0)
+## data.table 1.13.0 2020-07-24 [1] RSPM (R 4.0.2)
+## DBI 1.1.0 2019-12-15 [1] RSPM (R 4.0.0)
+## digest 0.6.25 2020-02-23 [1] RSPM (R 4.0.0)
+## DO.db 2.9 2020-11-25 [1] Bioconductor
+## DOSE 3.16.0 2020-10-27 [1] Bioconductor
+## downloader 0.4 2015-07-09 [1] RSPM (R 4.0.0)
+## dplyr 1.0.2 2020-08-18 [1] RSPM (R 4.0.2)
+## ellipsis 0.3.1 2020-05-15 [1] RSPM (R 4.0.0)
+## enrichplot 1.10.1 2020-11-14 [1] Bioconductor
+## evaluate 0.14 2019-05-28 [1] RSPM (R 4.0.0)
+## fansi 0.4.1 2020-01-08 [1] RSPM (R 4.0.0)
+## farver 2.0.3 2020-01-16 [1] RSPM (R 4.0.0)
+## fastmatch 1.1-0 2017-01-28 [1] RSPM (R 4.0.0)
+## fgsea 1.16.0 2020-10-27 [1] Bioconductor
+## generics 0.0.2 2018-11-29 [1] RSPM (R 4.0.0)
+## getopt 1.20.3 2019-03-22 [1] RSPM (R 4.0.0)
+## ggforce 0.3.2 2020-06-23 [1] RSPM (R 4.0.2)
+## ggplot2 3.3.2 2020-06-19 [1] RSPM (R 4.0.1)
+## ggraph 2.0.3 2020-05-20 [1] RSPM (R 4.0.2)
+## ggrepel 0.8.2 2020-03-08 [1] RSPM (R 4.0.2)
+## ggupset 0.3.0 2020-05-05 [1] RSPM (R 4.0.0)
+## glue 1.4.2 2020-08-27 [1] RSPM (R 4.0.2)
+## GO.db 3.12.1 2020-11-25 [1] Bioconductor
+## GOSemSim 2.16.1 2020-10-29 [1] Bioconductor
+## graphlayouts 0.7.0 2020-04-25 [1] RSPM (R 4.0.2)
+## gridExtra 2.3 2017-09-09 [1] RSPM (R 4.0.0)
+## gtable 0.3.0 2019-03-25 [1] RSPM (R 4.0.0)
+## hms 0.5.3 2020-01-08 [1] RSPM (R 4.0.0)
+## htmltools 0.5.0 2020-06-16 [1] RSPM (R 4.0.1)
+## igraph 1.2.6 2020-10-06 [1] RSPM (R 4.0.2)
+## IRanges * 2.24.0 2020-10-27 [1] Bioconductor
+## jsonlite 1.7.1 2020-09-07 [1] RSPM (R 4.0.2)
+## knitr 1.30 2020-09-22 [1] RSPM (R 4.0.2)
+## labeling 0.3 2014-08-23 [1] RSPM (R 4.0.0)
+## lattice 0.20-41 2020-04-02 [2] CRAN (R 4.0.2)
+## lifecycle 0.2.0 2020-03-06 [1] RSPM (R 4.0.0)
+## magrittr * 1.5 2014-11-22 [1] RSPM (R 4.0.0)
+## MASS 7.3-51.6 2020-04-26 [2] CRAN (R 4.0.2)
+## Matrix 1.2-18 2019-11-27 [2] CRAN (R 4.0.2)
+## memoise 1.1.0 2017-04-21 [1] RSPM (R 4.0.0)
+## msigdbr * 7.2.1 2020-10-02 [1] RSPM (R 4.0.2)
+## munsell 0.5.0 2018-06-12 [1] RSPM (R 4.0.0)
+## optparse * 1.6.6 2020-04-16 [1] RSPM (R 4.0.0)
+## org.Hs.eg.db * 3.12.0 2020-11-25 [1] Bioconductor
+## pillar 1.4.6 2020-07-10 [1] RSPM (R 4.0.2)
+## pkgconfig 2.0.3 2019-09-22 [1] RSPM (R 4.0.0)
+## plyr 1.8.6 2020-03-03 [1] RSPM (R 4.0.2)
+## polyclip 1.10-0 2019-03-14 [1] RSPM (R 4.0.0)
+## ps 1.4.0 2020-10-07 [1] RSPM (R 4.0.2)
+## purrr 0.3.4 2020-04-17 [1] RSPM (R 4.0.0)
+## qvalue 2.22.0 2020-10-27 [1] Bioconductor
+## R.cache 0.14.0 2019-12-06 [1] RSPM (R 4.0.0)
+## R.methodsS3 1.8.1 2020-08-26 [1] RSPM (R 4.0.2)
+## R.oo 1.24.0 2020-08-26 [1] RSPM (R 4.0.2)
+## R.utils 2.10.1 2020-08-26 [1] RSPM (R 4.0.2)
+## R6 2.4.1 2019-11-12 [1] RSPM (R 4.0.0)
+## RColorBrewer 1.1-2 2014-12-07 [1] RSPM (R 4.0.0)
+## Rcpp 1.0.5 2020-07-06 [1] RSPM (R 4.0.2)
+## readr 1.4.0 2020-10-05 [1] RSPM (R 4.0.2)
+## rematch2 2.1.2 2020-05-01 [1] RSPM (R 4.0.0)
+## reshape2 1.4.4 2020-04-09 [1] RSPM (R 4.0.2)
+## rlang 0.4.8 2020-10-08 [1] RSPM (R 4.0.2)
+## rmarkdown 2.4 2020-09-30 [1] RSPM (R 4.0.2)
+## RSQLite 2.2.1 2020-09-30 [1] RSPM (R 4.0.2)
+## rstudioapi 0.11 2020-02-07 [1] RSPM (R 4.0.0)
+## rvcheck 0.1.8 2020-03-01 [1] RSPM (R 4.0.0)
+## S4Vectors * 0.28.0 2020-10-27 [1] Bioconductor
+## scales 1.1.1 2020-05-11 [1] RSPM (R 4.0.0)
+## scatterpie 0.1.5 2020-09-09 [1] RSPM (R 4.0.2)
+## sessioninfo 1.1.1 2018-11-05 [1] RSPM (R 4.0.0)
+## shadowtext 0.0.7 2019-11-06 [1] RSPM (R 4.0.0)
+## stringi 1.5.3 2020-09-09 [1] RSPM (R 4.0.2)
+## stringr 1.4.0 2019-02-10 [1] RSPM (R 4.0.0)
+## styler 1.3.2 2020-02-23 [1] RSPM (R 4.0.0)
+## tibble 3.0.4 2020-10-12 [1] RSPM (R 4.0.2)
+## tidygraph 1.2.0 2020-05-12 [1] RSPM (R 4.0.2)
+## tidyr 1.1.2 2020-08-27 [1] RSPM (R 4.0.2)
+## tidyselect 1.1.0 2020-05-11 [1] RSPM (R 4.0.0)
+## tweenr 1.0.1 2018-12-14 [1] RSPM (R 4.0.2)
+## vctrs 0.3.4 2020-08-29 [1] RSPM (R 4.0.2)
+## viridis 0.5.1 2018-03-29 [1] RSPM (R 4.0.0)
+## viridisLite 0.3.0 2018-02-01 [1] RSPM (R 4.0.0)
+## withr 2.3.0 2020-09-22 [1] RSPM (R 4.0.2)
+## xfun 0.18 2020-09-29 [1] RSPM (R 4.0.2)
+## yaml 2.2.1 2020-02-01 [1] RSPM (R 4.0.0)
+##
+## [1] /usr/local/lib/R/site-library
+## [2] /usr/local/lib/R/libraryReferences
+Ahlmann-Eltze C., 2020 ggupset: Combination matrix axis for ’ggplot2’ to create ’upset’ plots. https://github.com/const-ae/ggupset
+Carlson M., 2019 Genome wide annotation for mouse. https://bioconductor.org/packages/release/data/annotation/html/org.Mm.eg.db.html
+Dolgalev I., 2020 Msigdbr: MSigDB gene sets for multiple organisms in a tidy data format. https://cran.r-project.org/web/packages/msigdbr/index.html
+Kanehisa M., and S. Goto, 2000 KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Research 28: 27–30. https://doi.org/10.1093/nar/28.1.27
+Khatri P., M. Sirota, and A. J. Butte, 2012 Ten years of pathway analysis: Current approaches and outstanding challenges. PLOS Computational Biology 8: e1002375. https://doi.org/10.1371/journal.pcbi.1002375
+Langfelder P., and S. Horvath, 2008 WGCNA: An r package for weighted correlation network analysis. BMC Bioinformatics 9. https://doi.org/10.1186/1471-2105-9-559
+Puthier D., and J. van Helden, 2015 Statistics for Bioinformatics - Practicals - Gene enrichment statistics. https://dputhier.github.io/ASG/practicals/go_statistics_td/go_statistics_td_2015.html
+Subramanian A., P. Tamayo, V. K. Mootha, S. Mukherjee, and B. L. Ebert et al., 2005 Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences 102: 15545–15550. https://doi.org/10.1073/pnas.0506580102
+Yaari G., C. R. Bolen, J. Thakar, and S. H. Kleinstein, 2013 Quantitative set analysis for gene expression: A method to quantify gene set differential expression including gene-gene correlations. Nucleic Acids Research 41: e170. https://doi.org/10.1093/nar/gkt660
+Yu G., L.-G. Wang, Y. Han, and Q.-Y. He, 2012 clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS: A Journal of Integrative Biology 16: 284–287. https://doi.org/10.1089/omi.2011.0118
+Yu G., clusterProfiler: Universal enrichment tool for functional and comparative study. http://yulab-smu.top/clusterProfiler-book/index.html
+