05. Peak calling and normalization
The pipeline requires a sample configuration file which provides information about ChIP-Input pairs and the type of peak calling to perform (broad or narrow).
This configuration file must be in a tab-delimited text file (including column names) containing the following information (respect the column order):
| target_id | input_id | broad |
|---|---|---|
| sample_A | input_A-B | False |
| sample_B | input_A-B | False |
| sample_C | input_C | True |
A dummy-table could be found in resources/peakCalling_sampleConfig_example.txt.
Despite many parameters are already available in the SPACCa_ChIPanalyses_configfile.yaml file, some additional information must be updated in the configuration file:
- the source bam directory (e.g. rename folder)
- the output directory where you want your results to be stored (if not already available, the pipeline will make it for you)
- whether your data contain UMIs
- whether your data are paired- or single-end
- the genome to use (in fasta format, and the genome id for CNV calling)
- the path to the sample configuration table
Hereafter there are some details of additional parameters available in the configfile_peakCalling.yaml. However, default parameters are already pre-set and should not be changed without expert advices.
If you wish to make changes, just make a copy of the config file and provide the path to the new file in the snakemake running command line.
| Parameter | Description |
|---|---|
| workflow_configuration: | |
| runs_directory | Full path (string) to the directory containing the bam files to be processed. |
| output_directory | Full path (string) to the directory where the pipeline outputs should be stored. |
| sample_config_table | Full path (string) to the sample configuration table. For details see the paragraph 5.1.1. |
| bam_features: | |
| bam_suffix | Default: ".bam". String with the suffix of the source bam files. |
| umi_present | Default: True. True/False to indicate whether the data contain UMIs (ignored for single-end data). |
| paired_end | Default: True. True/False to indicate whether the data are paired-end or not. |
| skip_bam_filtering | Default: False. True/False to indicate whether the bam MAPQ filtering and MarkDuplicates should be skipped. Useful to save computation when the bam files have been generated by the DNA-mapping pipeline. The bams will be linked in a folder, while the .bai index and the flagstat are re-computed. |
| remove_duplicates | Default: False. True/False to define whether remove the duplicates from the bam files (if true the tag in the bams will be _dedup instead of _mdup). |
| MAPQ_threshold | Default: 20. All reads with a mapping quality (MAPQ) score lower than this value will be filtered out from the bam files. |
| genomic_annotations: | |
| genome_id | No default. A string idicating the genome id (used only for the CNV correction). |
| genome_fasta | The full path to a fasta-formatted file containing the sequence of there reference genome into which the reads will be aligned. If the index (.fai) file is not present in the same folder, it will be build by the pipeline during its execution. The reference genomes can be downloaded, for instance, from the UCSC golden path. |
| blacklist | The full path to a BED-formatted file containing the regions corresponding to blacklisted regions (regions masked for normalization and peak calling). Blacklisted regions for the most common genome assemblies can be downloaded from the Boyle lab's git-page. |
| effective_genomeSize | Effective genome size used to normalize the ATAC-seq signal; e.g. for Hg38: 2900338458. A table for the most common genome assemblies is available in this repository at SPACCa/resources/chromosome_sizes_for_normalization.yaml. |
| ignore_for_normalization | A string of space separated chromosome names that should be excluded for ChIP-seq signal normalization, e.g. "X Y MT M KI270728.1". A table for the most common genome assemblies is available in this repository at SPACCa/resources/chromosome_sizes_for_normalization.yaml. |
| Other peakcalling parameters: | |
| bigWig_binSize | Default: 50. Size, in bp, of the bins used to compute the normalized bigWig files. |
| use_macs3 | Default: False. True/False to define whether to run macs3 instead of macs2. |
| macs_qValue_cutoff | Default: 0.01. False Discovery Ratio (FDR) (q-value) cutoff used by MACS to filter the significant peaks. |
| perform_plotFingerprint | Default: False. True/False to define whether perform the finger printing (Lorenz curve). |
| perform_fragmentSizeDistribution | Default: False. True/False to define whether to plot the fragment size distribution (Paired-end only). |
| fragment_length | Default: 200. Size in bp of the virtual fragment length at which each read should be extended in order to perform the plotFingerprint. |
| correlation_heatmap_colorMap | Default: 'PuBuGn'. A string indicating the color gradient pattern to use for the correlation heatmaps. This value is passed to matplotlib/seaborn. Therefore, available options (see matplotlib page for examples) are the following: 'Accent', 'Blues', 'BrBG', 'BuGn', 'BuPu', 'CMRmap', 'Dark2', 'GnBu', 'Greens', 'Greys', 'OrRd', 'Oranges', 'PRGn', 'Paired', 'Pastel1', 'Pastel2', 'PiYG', 'PuBu', 'PuBuGn', 'PuOr', 'PuRd', 'Purples', 'RdBu', 'RdGy', 'RdPu', 'RdYlBu', 'RdYlGn', 'Reds', 'Set1', 'Set2', 'Set3', 'Spectral', 'Wistia', 'YlGn', 'YlGnBu', 'YlOrBr', 'YlOrRd', 'afmhot', 'autumn', 'binary', 'bone', 'brg', 'bwr', 'cividis', 'cool', 'coolwarm', 'copper', 'cubehelix', 'flag', 'gist_earth', 'gist_gray', 'gist_heat', 'gist_ncar', 'gist_rainbow', 'gist_stern', 'gist_yarg', 'gnuplot', 'gnuplot2', 'gray', 'hot', 'hsv', 'icefire', 'inferno', 'jet', 'magma', 'mako', 'nipy_spectral', 'ocean', 'pink', 'plasma', 'prism', 'rainbow', 'rocket', 'seismic', 'spring', 'summer', 'tab10', 'tab20', 'tab20b', 'tab20c', 'terrain', 'twilight', 'twilight_shifted', 'viridis', 'vlag', 'winter'. |
To partially avoid unexpected errors during the execution of the pipeline, a so called 'dry-run' is strongly recommended. Indeed, adding a -n flag at the end of the snakemake running command will allow snakemake to check that all links and file/parameters dependencies are satisfied before to run the "real" processes. This command will therefore help the debugging process.
Always activate your environment, otherwise the pipeline won't be able to find the packages required for the analyses.
NOTE: if the bam files derive from the DNA-mapping pipeline you can save time by adding the flag
skip_bam_filtering="True"(MAPQ filter and MarkDuplicates are skipped). Notice that you may need to add/modify the flag for the bam suffix to something like"_mapq20_mdup_sorted.bam"in order to match the extension of the output files of the DNA-mapping pipeline.
snakemake \
--cores 10 \
-s </target/folder>/SPACCa/workflow/SPACCa_ChIPanalyses.snakefile \
--configfile </target/folder>/SPACCa/config/SPACCa_ChIPanalyses_configfile.yaml \
-nIf no errors occur, the pipeline can be run with the same command but without the final -n flag.
Notice that the absence of errors does not mean that the pipeline will run without any issues; the "dry-run" is only checking whether all the resources are available.
Here after you can see the full potential workflow of the paired-end and single-end ChIP-seq pipeline:
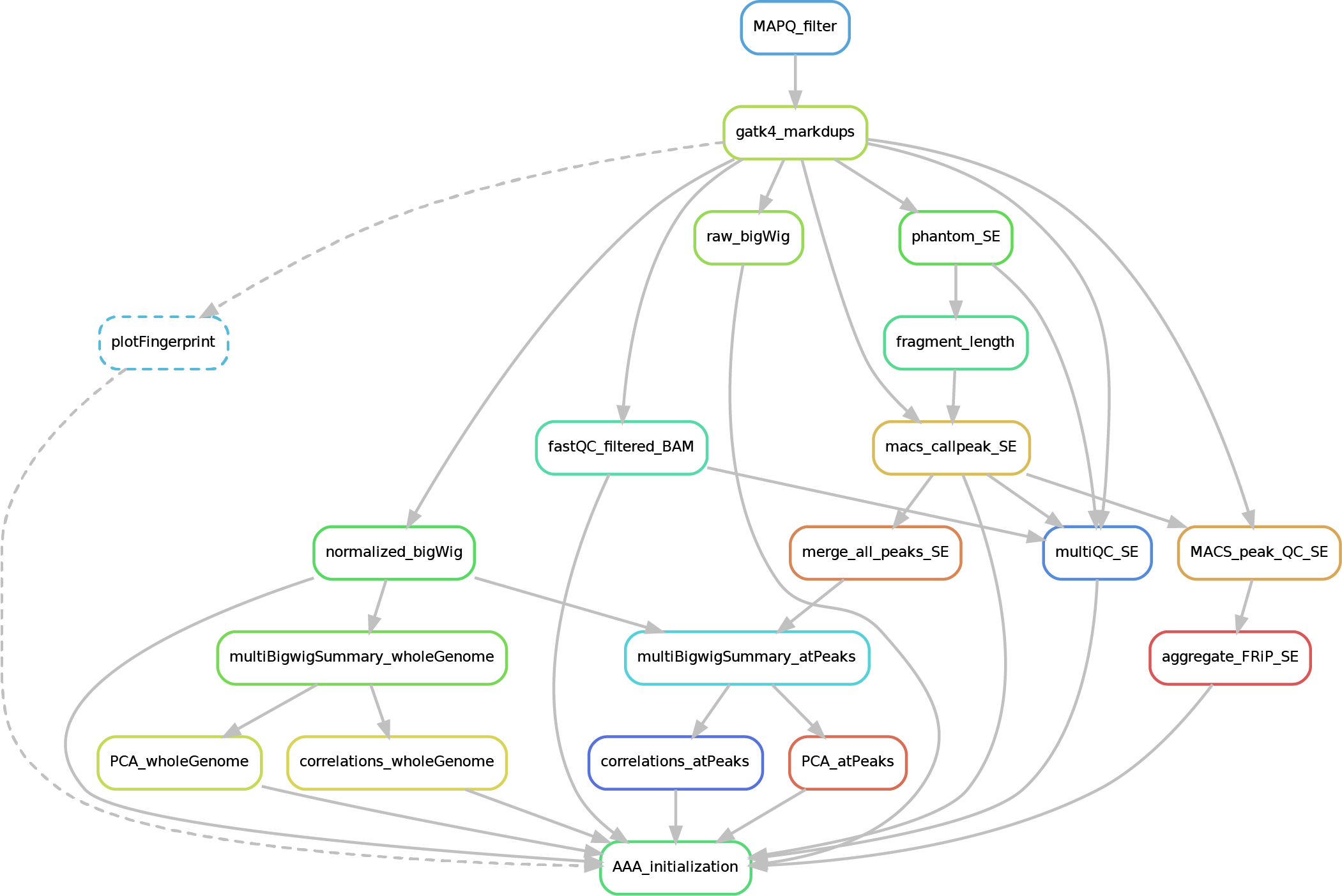
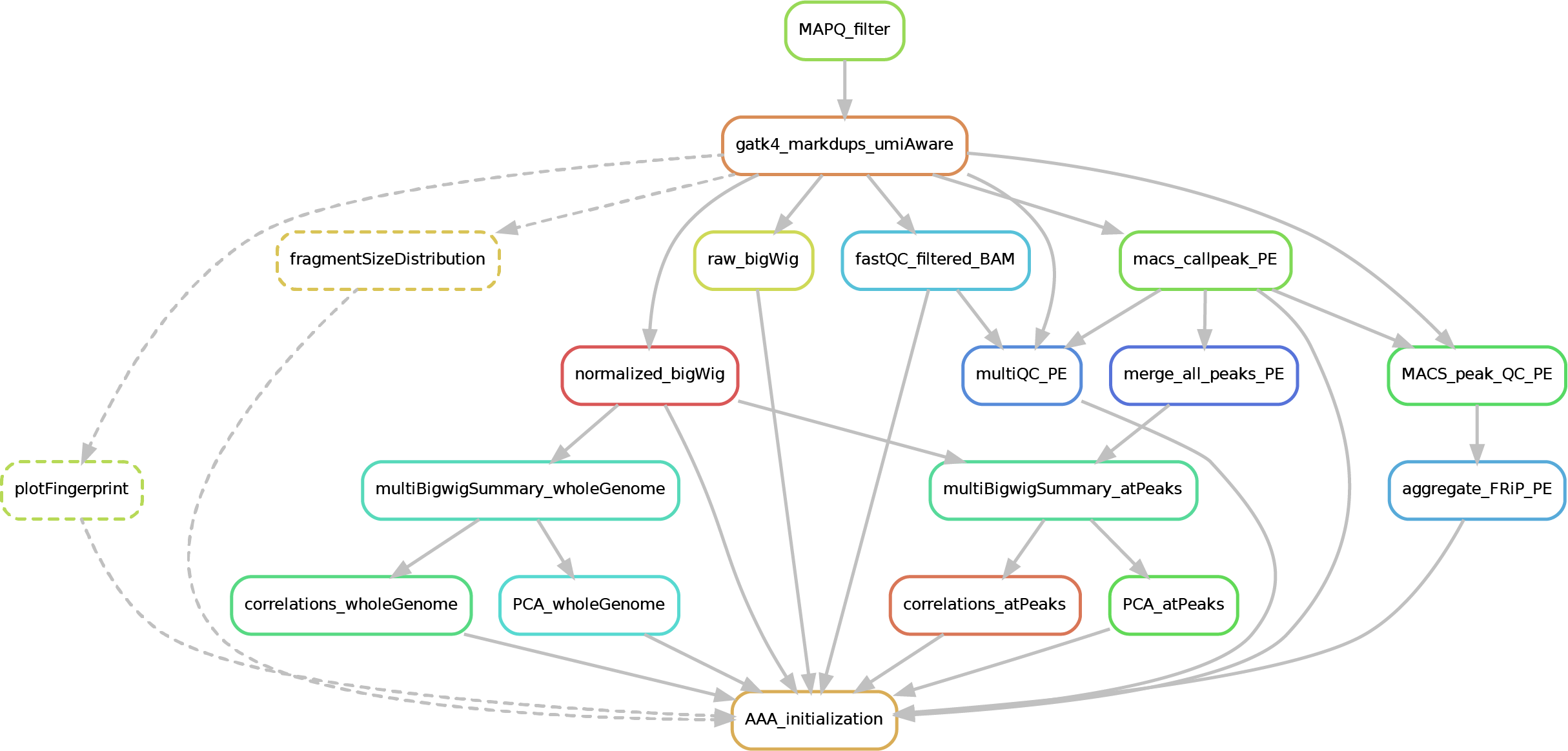
The results structure is the following:
- 01_BAM_filtered -> bams filtered for mapping quality (MAPQ) and with the duplicates marked/removed (if filtering was skipped, this folder will contain a symbolic link to the source bam and new indexes). If GCbias correction has been requested, a folder named GCbias_corrected_files will contain GC corrected bams (do not apply any deduplication on these files!)
- 02_fastQC_on_BAM_filtered -> individual fastQC for each filtered bam
- 03_bigWig_bamCoverage -> bigWig of the bam coverage normalized (RPGC = Reads Per Genomic Content) or not (raw_coverage) depending on the sequencing depth. If GC-bias and/or CNV have been requested, other sub-folders may be included: RPGC_normalized_CNA.corrected, RPGC_normalized_GCcorrected.
-
04_Called_peaks -> peaks called with macs2/3 (de-blacklisted). If single-end, it can be found also a folder with the output of
phantompeakqualtoolsas the calculated fragment length is used for running macs2/3 with single-end data. If the bed file does not contain already the 'chr' for the "canonical" chromosomes, it will be added in a separated file ending by_chr.narrow/broadPeak - 05_Quality_controls_and_statistics -> this folder contains sample correlations heatmaps and PCAs, a multiQC report containing multiple info (number of reads, duplicates, peak counts and fragmenth lenght, phantom results), statistics on the called peaks (FRiP, number, etc.)
The peak files generated by MACS2 (more info here) are composed by a canonical BED6 file (6-columns) and 4 extra columns:
Standard BED6
| Column | input_id | broad |
|---|---|---|
| 1 | chrom | Name of the chromosome (or contig, scaffold, etc.). |
| 2 | chromStart | The starting position of the feature in the chromosome or scaffold. The first base in a chromosome is numbered 0. |
| 3 | chromEnd | The ending position of the feature in the chromosome or scaffold. The chromEnd base is not included in the display of the feature. For example, the first 100 bases of a chromosome are defined aschromStart=0, chromEnd=100, and span the bases numbered 0-99. |
| 4 | name | Name given to a region (preferably unique). Use '.' if no name is assigned. |
| 5 | score | Indicates how dark the peak will be displayed in the browser (0-1000). If all scores were '0' when the data were submitted to the DCC, the DCC assigned scores 1-1000 based on signal value. Ideally the average signalValue per base spread is between 100-1000. |
| 6 | strand | +/- to denote strand or orientation (whenever applicable). Use '.' if no orientation is assigned. |
MACS2 specific columns
| 7 | signalValue | Measurement of overall (usually, average) enrichment for the region. |
| 8 | pValue | Measurement of statistical significance (-log10). Use -1 if no pValue is assigned. |
| 9 | qValue | Measurement of statistical significance using false discovery rate (-log10). Use -1 if no qValue is assigned. |
| 10 | peak | Point-source called for this peak; 0-based offset from chromStart. Use -1 if no point-source called. |
output_folder
├── 01_BAM_filtered
│ ├── sample_mapq20_mdup_sorted.bam
│ ├── sample_mapq20_mdup_sorted.bai
│ ├── flagstat
│ │ └── sample_mapq20_mdup_sorted_flagstat.txt
| ├── MarkDuplicates_metrics
│ │ └── sample_MarkDuplicates_metrics.txt
│ └── umi_metrics ### (if UMI present) ##
│ └── sample_UMI_metrics.txt
|
├── 02_fastQC_on_BAM_filtered
│ ├── sample_sorted_woMT_dedup_fastqc.html
│ └── sample_sorted_woMT_dedup_fastqc.zip
|
├── 03_bigWig_bamCoverage
│ ├── raw_coverage
│ │ └── sample_mapq20_mdup_raw.coverage_bs10.bw
│ └── RPGC_normalized
│ └── sample_mapq20_mdup_RPGC.normalized_bs10.bw
│
├── 04_Called_peaks
│ ├──phantom
│ │ └── sample.phantom.ssp.out
│ ├── sample.filtered.BAMPE_peaks_chr.narrowPeak ### .BAM if Single-End ###
│ ├── sample.filtered.BAMPE_peaks.narrowPeak ### .BAM if Single-End ###
│ └── sample.filtered.BAMPE_peaks.xls ### .BAM if Single-End ###
|
└── 05_Quality_controls_and_statistics
├── multiQC
│ └── multiQC_report.html
├── peaks_stats
│ └── all_samples_FRiP_report.tsv
├── plotFingerprint ### optional ###
| ├── quality_metrics
| │ └── sample_fingerPrinting_quality_metrics.txt
| └── sample_fingerPrinting_plot.pdf
├── sample_comparisons_atPeaks
│ ├── all_peaks_merged_sorted.bed
│ ├── multiBigWigSummary_matrix_atPeaks.npz
│ ├── sample_pearson.correlation_heatmap_atPeaks.pdf
│ ├── sample_spearman.correlation_heatmap_atPeaks.pdf
│ ├── sample_correlation_PCA.1-2_heatmap_atPeaks.pdf
│ └── sample_correlation_PCA.2-3_heatmap_atPeaks.pdf
└── sample_comparisons_wholeGenome
├── all_peaks_merged_sorted.bed
├── multiBigWigSummary_matrix_atPeaks_inputs.npz
├── multiBigWigSummary_matrix_atPeaks_targets.npz
├── sample_pearson.correlation_heatmap_wholeGenome_inputs.pdf
├── sample_pearson.correlation_heatmap_wholeGenome_targets.pdf
├── sample_spearman.correlation_heatmap_wholeGenome_inputs.pdf
├── sample_spearman.correlation_heatmap_wholeGenome_targets.pdf
├── sample_correlation_PCA.1-2_heatmap_wholeGenome_inputs.pdf
├── sample_correlation_PCA.1-2_heatmap_wholeGenome_targets.pdf
├── sample_correlation_PCA.2-3_heatmap_wholeGenome_inputs.pdf
└── sample_correlation_PCA.2-3_heatmap_wholeGenome_targets.pdf
Contributors