

ToulligQC is dedicated to the QC analyses of Oxford Nanopore runs. This software is written in Python and developped by the GenomiqueENS core facility of the Institute of Biology of the Ecole Normale Superieure (IBENS).
Click on following image to see an report example. An online help is available to better understand graphics generated with ToulligQC when clicking on the ⓘ icon.
Karine Dias, Bérengère Laffay, Lionel Ferrato-Berberian, Sophie Lemoine, Ali Hamraoui, Morgane Thomas-Chollier, Stéphane Le Crom and Laurent Jourdren.
Support is availlable on GitHub issue page and at toulligqc at bio.ens.psl.eu.
-
2.Usage
- 2.1 Command line
- 2.2 Sample data
-
3.Output
This option is also suitable if you are interested in further developments of the package, but requires a little bit more hands-on. Install the dependencies required and clone the repository locally.
$ git clone https://github.com/GenomicParisCentre/toulligQC.git
# X.X here is the version of ToulligQC to install
$ git checkout vX.X
$ cd toulligqc && python3 setup.py build install- Requirements
ToulligQC is written with Python 3. To run ToulligQC without Docker, you need to install the following Python modules:
- matplotlib
- plotly
- h5py
- pandas
- numpy
- scipy
- scikit-learn
- pysam
- tqdm
- pod5
You can use a conda environment to install the required packages:
git clone https://github.com/GenomicParisCentre/toulligQC.git
cd toulligqc && python3 setup.py build install
conda env create -f environment.yml
conda activate toulliqc
ToulligQC can be more easlily installed with a pip package availlable on the PyPi repository. The following command line will install the latest version of ToulligQC:
$ pip3 install toulligqcToulligQC and its dependencies are available through a Docker image. To install docker on your system, go to the Docker website (https://docs.docker.com/engine/installation/). Even if Docker can run on Windows or macOS virtual machines, we recommend to run ToulligQC on a Linux host.
An image of ToulligQC is hosted on the Docker hub on the genomicpariscentre repository (genomicpariscentre/toulligqc).
$ docker pull genomicpariscentre/toulligqc:latest$ docker run -ti \
-u $(id -u):$(id -g) \
--rm \
-v /path/to/basecaller/sequencing/summary/file:/path/to/basecaller/sequencing/summary/file \
-v /path/to/basecaller/sequencing/telemetry/file:/path/to/basecaller/telemetry/summary/file \
-v /path/to/result/directory:/path/to/result/directory \
genomicpariscentre/toulligqc:latest
ToulligQC is also available on nf-core as a module written in nextflow. To install nf-core on your system, please visit their website (https://nf-co.re/docs/usage/introduction).
The following command line will install the latest version of the ToulligQC module:
$ nf-core modules install toulligqcToulligQC is adapted to RNA-Seq along with DNA-Seq and it is compatible with 1D² runs. This QC tool supports only Guppy and Dorado basecalling ouput files. It also needs a single FAST5 file (to catch the flowcell ID and the run date) if a telemetry file is not provided. Flow cells and kits version are retrieved using the telemetry file. ToulligQC can take barcoding samples by adding the barcode list as a command line option.
If the sequencing summary file is not available, toulligQC can also accept FASTQ or BAM files.
To do so, ToulligQC deals with different file formats: gz, tar.gz, bz2, tar.bz2 and .fast5 to retrieve a FAST5 information. This tool will produce a set of graphs, statistic file in plain text format and a HTML report.
To run ToulligQC you need the Guppy/ Dorado basecaller output files : sequencing_summary.txt and sequencing_telemetry.js. or FASTQ or BAM
This can be compressed with gzip or bzip2.
You can use your initial Fast5 ONT file too.
ToulligQC can perform analyses on your data if the directory is organised as the following:
RUN_ID
├── sequencing_summary.txt
└── sequencing_telemetry.js
for 1D² analysis:
RUN_ID
├── sequencing_summary.txt
├── sequencing_telemetry.js
└── 1dsq_analysis
└── sequencing_1dsq_summary.txt
For a barcoded run you can add the barcoding files generated by Guppy/ Dorado barcoding_summary_pass.txt and barcoding_summary_fail.txt to ToulligQC or a single file sequencing_summary_all.txt containing sequencing_summary and barcoding_summary information combined.
For the barcode list to use in the command line options, ToulligQC handle the following naming schemes: BCXX, RBXX, NBXX and barcodeXX where XX is the number of the barcode. The barcode naming schemes are case insensitive.
This is a directory for 1D² analysis with barcoding files:
RUN_ID
├── sequencing_summary.txt
├── sequencing_telemetry.js
└── 1dsq_analysis
├── barcoding_summary_pass.txt
├── barcoding_summary_fail.txt
└── sequencing_1dsq_summary.txt
General Options:
usage: ToulligQC V2.6 [-a SEQUENCING_SUMMARY_SOURCE] [-t TELEMETRY_SOURCE]
[-f FAST5_SOURCE] [-p POD5_SOURCE] [-q FASTQ] [-u BAM]
[--thread THREAD] [--batch-size BATCH_SIZE] [--qscore-threshold THRESHOLD]
[-n REPORT_NAME] [--output-directory OUTPUT] [-o HTML_REPORT_PATH]
[--data-report-path DATA_REPORT_PATH]
[--images-directory IMAGES_DIRECTORY]
[-d SEQUENCING_SUMMARY_1DSQR_SOURCE]
[-s SAMPLESHEET]
[-b] [-l BARCODES]
[--quiet] [--force] [-h] [--version]
required arguments:
-a SEQUENCING_SUMMARY_SOURCE, --sequencing-summary-source SEQUENCING_SUMMARY_SOURCE
Basecaller sequencing summary source, can be
compressed with gzip (.gz) or bzip2 (.bz2)
-t TELEMETRY_SOURCE, --telemetry-source TELEMETRY_SOURCE
Basecaller telemetry file source, can be compressed
with gzip (.gz) or bzip2 (.bz2)
-f FAST5_SOURCE, --fast5-source FAST5_SOURCE
Fast5 file source (necessary if no telemetry file),
can also be in a tar.gz/tar.bz2 archive or a directory
-p POD5_SOURCE, --pod5-source POD5_SOURCE
pod5 file source (necessary if no telemetry file),
can also be in a tar.gz/tar.bz2 archive or a directory
-q FASTQ, --fastq FASTQ
FASTQ file (necessary if no sequencing summary file),
can also be in a .gz archive
-u BAM, --bam BAM
BAM file (necessary if no sequencing summary file),
can also be a SAM format
optional arguments:
-s SAMPLESHEET, --samplesheet SAMPLESHEET
Samplesheet (.csv file) to fill out sample names in MinKNOW.
-n REPORT_NAME, --report-name REPORT_NAME
Report name
--output-directory OUTPUT
Output directory
-o HTML_REPORT_PATH, --html-report-path HTML_REPORT_PATH
Output HTML report
--data-report-path DATA_REPORT_PATH
Output data report
--images-directory IMAGES_DIRECTORY
Images directory
-d SEQUENCING_SUMMARY_1DSQR_SOURCE, --sequencing-summary-1dsqr-source SEQUENCING_SUMMARY_1DSQR_SOURCE
Basecaller 1dsq summary source
-b, --barcoding Option for barcode usage
-l BARCODES, --barcodes BARCODES
Comma-separated barcode list (e.g.,
BC05,RB09,NB01,barcode10) or a range separated with ':' (e.g.,
barcode01:barcode19)
--thread THREAD Number of threads for parsing FASTQ or BAM files (default: 2).
--batch-size BATCH_SIZE Batch size for each threads (default: 500).
--qscore-threshold THRESHOLD Q-score threshold to distinguish between passing filter and
fail reads (default: 9), applicable only for FASTQ and BAM files.
--quiet Quiet mode
--force Force overwriting of existing files
-h, --help Show this help message and exit
--version show program's version number and exit
-
Sequencing summary alone
Note that the fowcell ID and run date will be missing from report, found in telemetry file or single fast5 file
$ toulligqc --report-name summary_only \
--sequencing-summary-source /path/to/basecaller/output/sequencing_summary.txt \
--html-report-path /path/to/output/report.html- Sequencing summary + telemetry file
$ toulligqc --report-name summary_plus_telemetry \
--telemetry-source /path/to/basecaller/output/sequencing_telemetry.js \
--sequencing-summary-source /path/to/basecaller/output/sequencing_summary.txt \
--html-report-path /path/to/output/report.html- Telemetry file + fast5 files
$ toulligqc --report-name telemetry_plus_fast5 \
--telemetry-source /path/to/basecaller/output/sequencing_telemetry.js \
--fast5-source /path/to/basecaller/output/fast5_files.fast5.gz \
--html-report-path /path/to/output/report.html- Fastq/ bam files only
$ toulligqc --report-name FAF0256 \
--fastq /path/to/basecaller/output/fastq_files.fq.gz \ # (replace with --bam)
--html-report-path /path/to/output/report.html- Optional arguments for 1D² analysis
$ toulligqc --report-name FAF0256 \
--telemetry-source /path/to/basecaller/output/sequencing_telemetry.js \
--sequencing-summary-source /path/to/basecaller/output/sequencing_summary.txt \
--sequencing-summary-1dsqr-source /path/to/basecaller/output/sequencing_1dsqr_summary.txt \ # (optional)
--html-report-path /path/to/output/report.html- Optional arguments to deal with barcoded samples
$ toulligqc --report-name FAF0256 \
--barcoding \
--telemetry-source /path/to/basecaller/output/sequencing_telemetry.js \
--sequencing-summary-source /path/to/basecaller/output/sequencing_summary.txt \
--sequencing-summary-source /path/to/basecaller/output/barcoding_summary_pass.txt \ # (optional)
--sequencing-summary-source /path/to/basecaller/output/barcoding_summary_fail.txt \ # (optional)
--sequencing-summary-1dsqr-source /path/to/basecaller/output/sequencing_1dsqr_summary.txt \ # (optional)
--sequencing-summary-1dsqr-source /path/to/basecaller/output/barcoding_summary_pass.txt \ # (optional)
--sequencing-summary-1dsqr-source /path/to/basecaller/output/barcoding_summary_fail.txt \ # (optional)
--html-report-path /path/to/output/report.html \
--data-report-path /path/to/output/report.data \ # (optional)
--barcodes BC01,BC02,BC03We provide sample raw data that can be used to launch and evaluate our software. This demo data has been generated using a MinION MKIb with a R9.4.1 flowcell (FLO-MIN106) in 1D (SQK-LSK108) mode with barcoded samples (BC01, BC02, BC03, BC04, BC05 and BC07). Data acquisition was performed using MinKNOW 1.11.5 and basecalling/demultiplexing was completed using Guppy 3.2.4.
- First download and uncompress sample data:
$ wget http://outils.genomique.biologie.ens.fr/leburon/downloads/toulligqc-example/toulligqc_demo_data.tar.bz2
$ tar -xzf toulligqc_demo_data.tar.bz2
$ cd toulligqc_demo_data- Then, you can launch the ToulligQC analysis of the demo data with the
run-toulligqc-with-docker.shscript if you want to use a Docker container:
$ ./run-toulligqc-with-docker.sh- Or with
run-toulligqc.shscript if ToulligQC is already installed on your system:
$ ./run-toulligqc.sh- Of course, you can also launch manually ToulligQC on the sample data with the following command line:
$ toulligqc \
--report-name 'ToulligQC Demo Data' \
--barcoding \
--telemetry-source sequencing_telemetry.js \
--sequencing-summary-source sequencing_summary.txt \
--sequencing-summary-source barcoding_summary_pass.txt \
--sequencing-summary-source barcoding_summary_fail.txt \
--barcodes BC01:BC07 \
--output-directory outputWith this scripts or command line, ToulligQC will create an output directory with output HTML report.
More information about this sample data and scripts can be found in the README file of the tar archive.
If the options --output-directory or --html-report-path are not provided, ToulligQC generates all below files and images in the current directory.
If no report-name is given, ToulligQC creates a default report name.
-
A HTML report with (the path of this file can be defined using
--html-report-pathcommand line option ):- useful information about the sequencing run given as input
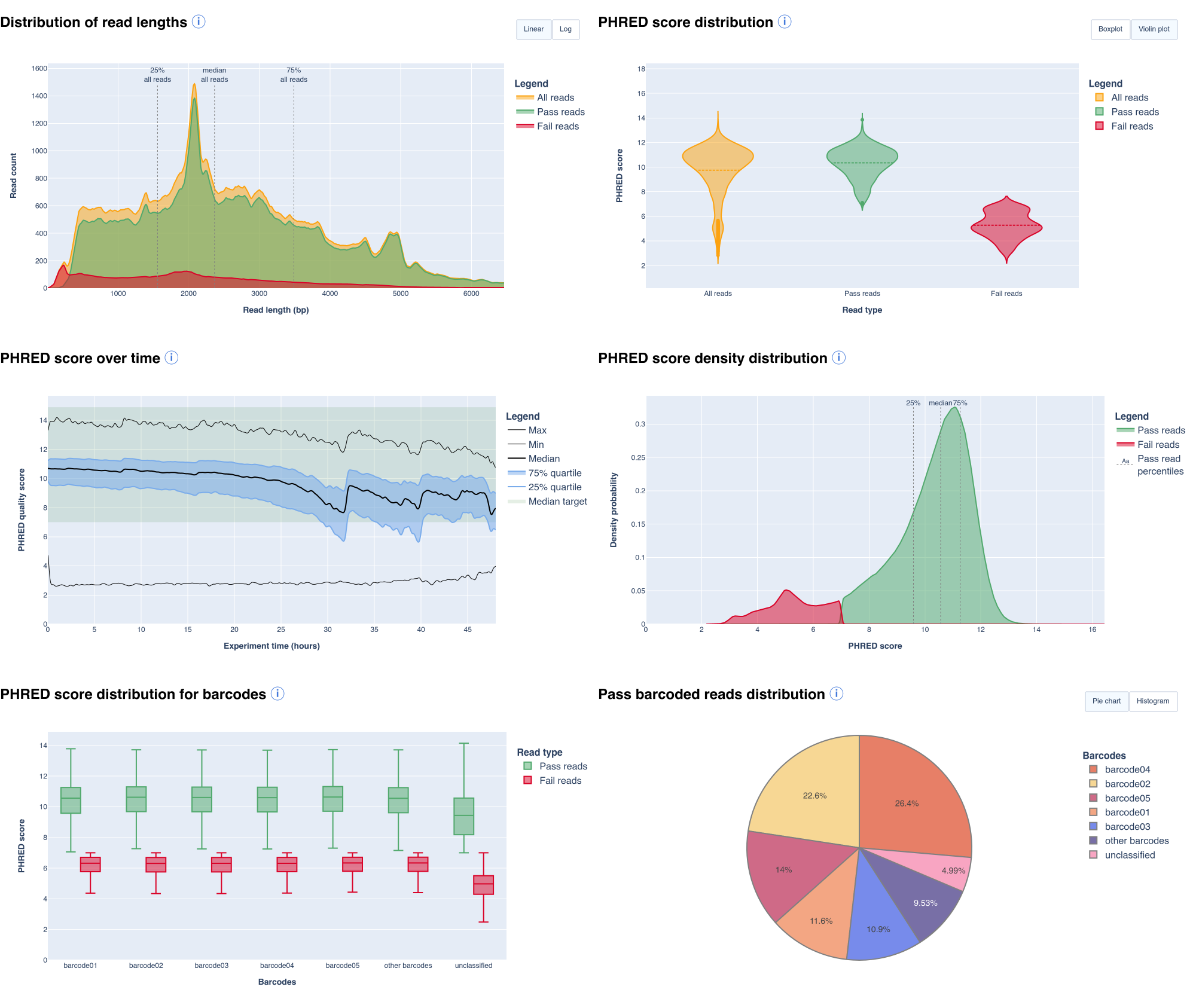
- a read count and a read length histograms about different read types
- a graph checking that the sequencing was homogeneous during a run
- a graph allowing to locate potential flowcell spatial biaises
- graphs representing the PHRED score distribution and the density distribution across read types
- a collection of graphs displaying length/speed/quality or number of sequences over sequencing time
- a set of graphs providing quality, length information and read counts for each barcode
-
A report.data log file containing (the path of this file can be defined using
--data-report-pathcommand line option ):- information about ToulligQC execution
- environment variables
- full statistics are provided for complementary analyses if needed : the information by modules is retained in a key-value form, the prefix of a key being the report data file id of the module
- the nucleotide rate per read
If you choose to use a directory output (default choice), the output will be organised like this :
RUN_ID
├── report.html
├── report.data
└── images
└── plots.html
└── plot.png