StellarPath is a deep learning patient classifier which leverages state-of-the-art bioinformatics techniques to select biologically meaningful molecular and pathway features, the pathway space to integrate different omics and increase the software interpretability, the PSN paradigm to enable analysis and stratification of patients based on their pathway activity, and an artificial neural network to account for the similarity information and classify.
StellarPath: hierarchical-vertical multi-omics classifier synergizes stable markers and interpretable similarity networks for patient profiling. Authors: Giudice Luca, Mohamed Ahmed and Malm Tarja
- Free of hyper-parameters (input data are the same of common pathway analysis tools)
- Analyses raw counts from their normalization to integration in pathways without need of user’s pre-processing (at the end, the user will have access also to the normalized counts)
- Integrates multi-omics information in pathways to ease interpretability and not deviate from biology
- Uses a novel measure to determine how much two patients are similar. It considers how much and in which direction two patients regulate a specific pathway.
- Creates pathway-specific PSNs where each node represents a patient in study, an edge connects two patients based on how much they are similar in a pathway.
- Select molecules and pathways if their PSNs satisfy a criterium (one class has patients who are more similar than the members of the opposite class and the two classes are not similar, i.e. one class is cohesive and the other one is sparse).
- Feeds the PSNs to a graph convolutional network that learns how to recognize the patients based on their similarities and how to predict the class of unseen individuals.
- PSNs can be visualized with an ad-hoc implementation that reduces the number of patients which are visible as nodes without losing their information.
- molecular markers (genes, miRNAs, …) which are differentially expressed, differentially stable and predictive of a patient’s class.
- pathways that are enriched by markers, significantly deregulated, represented by biologically meaningful PSNs, and predictive of a patient’s class.
- significant PSNs showing that the pathway activity of the patients of one class is precisely regulated with highly (positive/negative) stable molecules while the members of the opposite class present more variable and different expression levels.
- patient’s centrality score reflects how much each patient in study is similar to the members of its class and dissimilar from the others in a pathway. This enables the patient stratification.
We tested the installation and our package with R 3 and 4 both on Ubuntu 18/22 and Windows 11.
Functions to check and install (if necessary) and update (if necessary) a package
check_install <- function(package_name) {
if (!require(package_name, character.only = TRUE)) {
install.packages(package_name)
library(package_name, character.only = TRUE)
cat(sprintf("%s has been installed.\n", package_name))
} else {
cat(sprintf("%s is already installed.\n", package_name))
}
available_updates <- old.packages(lib.loc = .libPaths())
if (!is.null(available_updates) && package_name %in% rownames(available_updates)) {
update.packages(oldPkgs = package_name, ask = FALSE)
cat(sprintf("%s has been updated to the latest version.\n", package_name))
} else {
cat(sprintf("%s is up to date.\n", package_name))
}
}These packages need to be installed and updated to deploy StellarPath correctly
check_install("devtools")
check_install("BiocManager")
# This package needs to be installed manually
install.packages("https://bioconductor.org/packages/3.17/bioc/src/contrib/miRBaseConverter_1.24.0.tar.gz", repos = NULL, type = "source")This is the fastest way to install StellarPath, including all dependencies:
devtools::install_github(
repo="LucaGiudice/StellarPath",
ref="main",
dependencies = TRUE,
build_vignettes = FALSE
)- At the head of the github repository click: Code -> Download Zip
- Extract the Zip and Get the path of the directory like this: "C:\Users\Luca\Downloads\StellarPath-main"
- Run the following R code (in Windows the slash must be inverted)
setwd("C:/Users/Luca/Downloads/StellarPath-main")
install.packages(".", repos = NULL, type="source", dependencies = TRUE)Once the R package is installed, you can install the Python module. We recommend having Conda installed in the system but shouldn't be necessary. In case you would like to install Conda (and control the environment that StellarPath is going to install), we recommend using Conda Navigator which is very easy to install and use. The next operations with our package will install a Conda environment automatically. The Conda environment is needed to use graph convolutional neural networks which have been implemented in Python.
library("StellarPath")
SP_install() #check conda, if conda is missing installs automatically miniconda, then it installs the conda enviroment
SP_initialize(save_profile = TRUE) # activate the conda enviroment that is needed to use the packageThe installation is completed, you can run StellarPath like this:
library("StellarPath")
SP_initialize()In case of any installation issue:
We suggest to use the docker: lgiudice/py3_r4_bioinf:latest. It has an Ubuntu OS that is suited to install and work with StellarPath.
There are the following vignettes:
- Introduction to StellarPath's cross-validation workflow, data and results
- Second application and visualization of results
- StellarPath's inference with CLL data
- StellarPath's pathway categorization and similarity measure
- StellarPath vs GSEA
- StellarPath's inference with AD data
- From sequencing data in fasta to count matrix tutorial
- From mutation data in VCF to BIN matrix tutorial
In case of running the vignettes with Windows:
Remove everything related to "runningTime" because Windows does not allow measuring the running time and memory usage from an R script
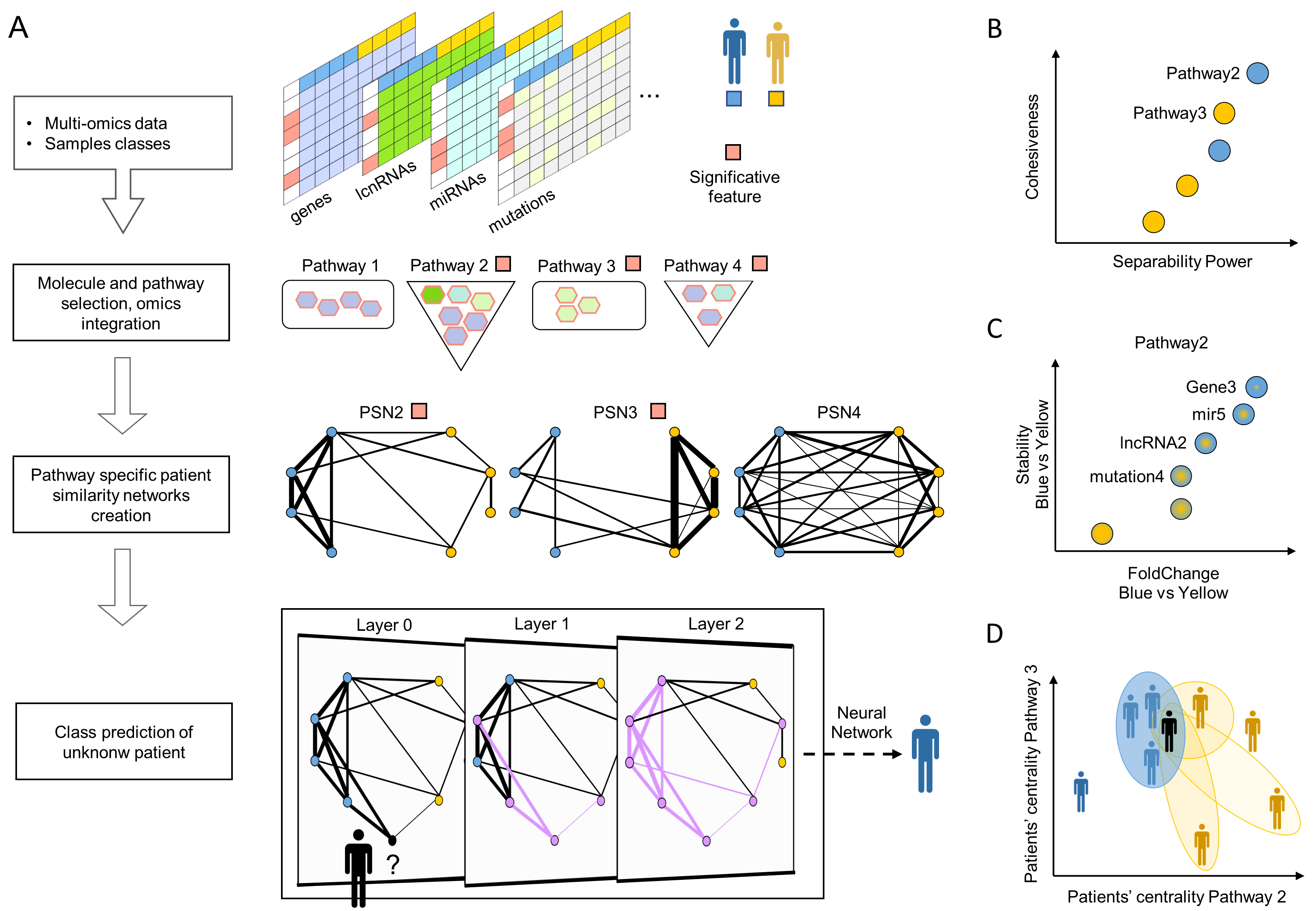
Frame A shows the workflow:
- The method classifies two patients’ classes and works with both dense and sparse (e.g., somatic mutation) omics.
- It finds the significantly deregulated molecules and their enriched pathways.
- It determines how much each pair of patients is similar by comparing the values of the molecules belonging to a specific pathway. It uses pathway-specific similarities to build a network. Only the PSNs, which show that one class is cohesive while the opposite one is not, are kept.
- StellarPath receives an unknown patient, adds it to a significant PSN and predicts its class with a graph convolutional network. The latter step is repeated with all PSNs and the final prediction is made by consensus/major voting.
- StellarPath provides multiple output data: B) a PSN represents the pathway’s similarities due to differentially expressed and variant molecules, C) the enriched pathways are prioritized with the properties of their PSNs, D) both known and unknown patients are stratified, and quality checked based on how much are similar to the others in the enriched pathways.
StellarPath integrates omics with a knowledge-based approach in pathways and takes charge of both the ome's processing and results. This means that, before putting new ome available in the system, we will test the results in wet lab to be sure that its integration with other data is not creating false biological findings. The current omics and supported integration is the following one:
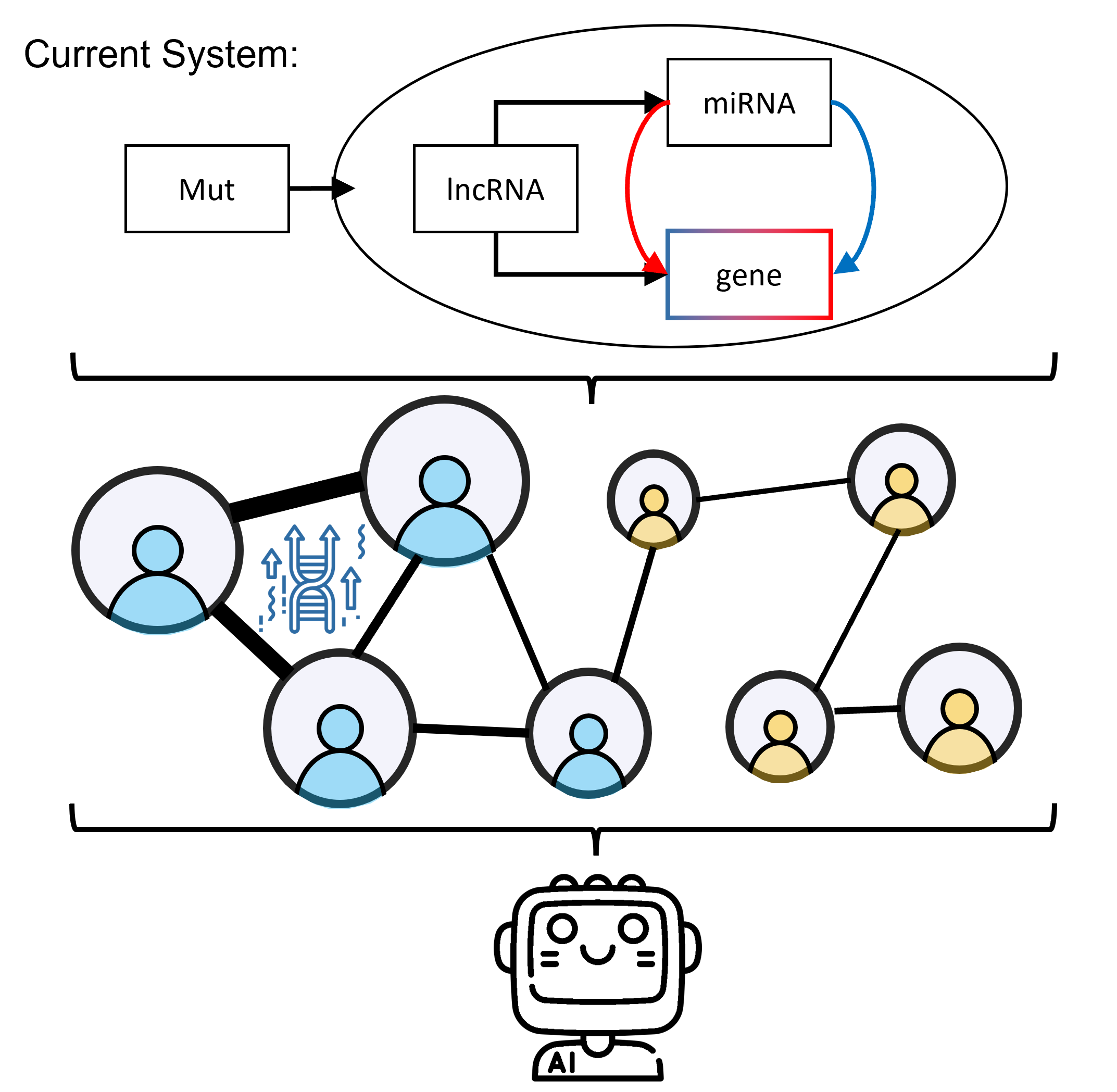
We will add other omics in the near future.
BSD-3-Clause @ Giudice Luca