![]()




![]()
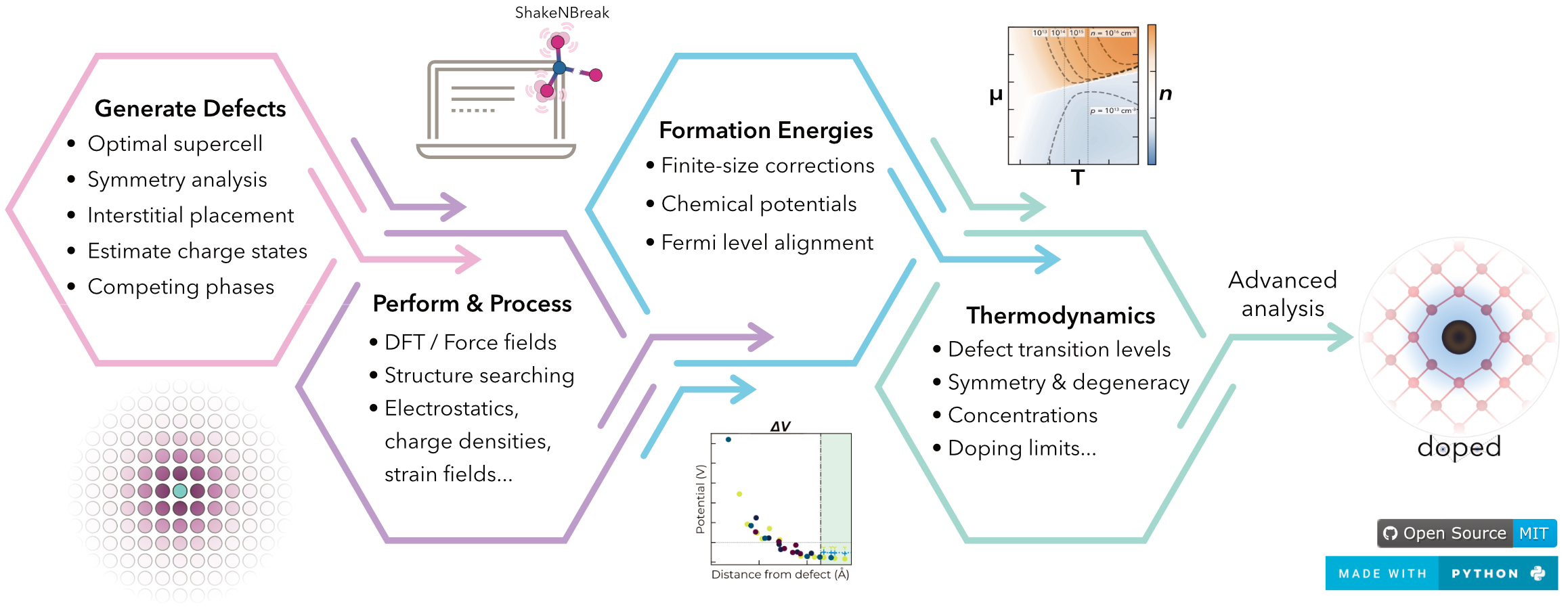
doped is a Python software for the generation, pre-/post-processing and analysis of defect supercell calculations, implementing the defect simulation workflow in an efficient, reproducible, user-friendly yet powerful and fully-customisable manner.
Tutorials showing the code functionality and usage are provided on the docs site, and an overview of the key advances of the package is given in the JOSS paper.
All features and functionality are fully-customisable:
- Supercell Generation: Generate an optimal supercell, maximising periodic image separation for the minimum number of atoms (computational cost).
- Defect Generation: Generate defect supercells and guess likely charge states based on chemical intuition.
- Calculation I/O: Automatically write inputs and parse calculations (
VASP& other DFT/force-field codes). - Chemical Potentials: Determine relevant competing phases for chemical potential limits, with automated calculation setup, parsing and analysis.
- Defect Analysis: Automatically parse calculation outputs to compute defect formation energies, finite-size corrections (FNV & eFNV), symmetries, degeneracies, transition levels, etc.
- Thermodynamic Analysis: Compute (non-)equilibrium Fermi levels, defect/carrier concentrations etc. as functions of annealing/cooling temperature, chemical potentials etc.
- Plotting: Generate publication-quality plots of defect formation energies, chemical potential limits, defect/carrier concentrations, Fermi levels, charge corrections, etc.
PythonInterface: Fully-customisable, modularPythonAPI. Plug-and-play w/ShakeNBreak– defect structure-searching,easyunfold– band unfolding,CarrierCapture.jl/nonrad– non-radiative recombination etc.- Reproducibility, tabulation, automated compatibility/sanity checking, strain/displacement analysis, shallow defect analysis, high-throughput compatibility, Wyckoff analysis...
 (a) Optimal supercell generation comparison. (b) Charge state estimation comparison. Example (c) Kumagai-Oba (eFNV) finite-size correction plot, (d) defect formation energy diagram, (e) chemical potential / stability region, (f) Fermi level vs. annealing temperature, (g) defect/carrier concentrations vs. annealing temperature and (h) Fermi level / carrier concentration heatmap plots from
(a) Optimal supercell generation comparison. (b) Charge state estimation comparison. Example (c) Kumagai-Oba (eFNV) finite-size correction plot, (d) defect formation energy diagram, (e) chemical potential / stability region, (f) Fermi level vs. annealing temperature, (g) defect/carrier concentrations vs. annealing temperature and (h) Fermi level / carrier concentration heatmap plots from doped. Automated plots of (i,j) single-particle eigenvalues and (k) site
displacements from DFT supercell calculations. See the JOSS paper for more details.
pip install doped # install doped and dependencies
conda install -c conda-forge spglib # bundle C libraries with spglibNote that either conda install -c conda-forge spglib or
pip install git+https://github.com/spglib/spglib --config-settings=cmake.define.SPGLIB_SHARED_LIBS=OFF
should be used after pip install doped, which ensures that the correct C libraries are bundled with
spglib, to prevent unnecessary warnings.
Alternatively if desired, doped can also be installed from conda with:
conda install -c conda-forge doped
pip install pydefect # pydefect not available on conda, so needs to be installed with pip or otherwise, if using the eFNV correctionIf you haven't done so already, you will need to set up your VASP POTCAR files and Materials Project API with pymatgen using the .pmgrc.yaml file, in order for doped to automatically generate VASP input files for defect calculations and determine competing phases for chemical potentials.
See the docs Installation page for details on this.
If you use doped in your research, please cite:
- S. R. Kavanagh et al. doped: Python toolkit for robust and repeatable charged defect supercell calculations. Journal of Open Source Software 9 (96), 6433, 2024
As shown in the doped tutorials, it is highly recommended to use the ShakeNBreak approach when calculating point defects in solids, to ensure you have identified the groundstate structures of your defects. As detailed in the theory paper, skipping this step can result in drastically incorrect formation energies, transition levels, carrier capture (basically any property associated with defects). This approach is followed in the doped example notebook, with a more in-depth explanation and tutorial given on the ShakeNBreak website.

- B. E. Murdock et al. Li-Site Defects Induce Formation of Li-Rich Impurity Phases: Implications for Charge Distribution and Performance of LiNi0.5-xMxMn1.5O4 Cathodes (M = Fe and Mg; x = 0.05–0.2) Advanced Materials 2024
- A. G. Squires et al. Oxygen dimerization as a defect-driven process in bulk LiNiO22 ChemRxiv 2024
- Y. Fu & H. Lohan et al. Factors Enabling Delocalized Charge-Carriers in Pnictogen-Based Solar Absorbers: In-depth Investigation into CuSbSe2 arXiv 2024
- S. Hachmioune et al. Exploring the Thermoelectric Potential of MgB4: Electronic Band Structure, Transport Properties, and Defect Chemistry Chemistry of Materials 2024
- J. Hu et al. Enabling ionic transport in Li3AlP2 the roles of defects and disorder ChemRxiv 2024
- X. Wang et al. Upper efficiency limit of Sb2Se3 solar cells Joule 2024
- I. Mosquera-Lois et al. Machine-learning structural reconstructions for accelerated point defect calculations npj Computational Materials 2024
- W. Dou et al. Band Degeneracy and Anisotropy Enhances Thermoelectric Performance from Sb2Si2Te6 to Sc2Si2Te6 Journal of the American Chemical Society 2024
- K. Li et al. Computational Prediction of an Antimony-based n-type Transparent Conducting Oxide: F-doped Sb2O5 Chemistry of Materials 2023
- X. Wang et al. Four-electron negative-U vacancy defects in antimony selenide Physical Review B 2023
- Y. Kumagai et al. Alkali Mono-Pnictides: A New Class of Photovoltaic Materials by Element Mutation PRX Energy 2023
- S. M. Liga & S. R. Kavanagh, A. Walsh, D. O. Scanlon, G. Konstantatos Mixed-Cation Vacancy-Ordered Perovskites (Cs2Ti1–xSnxX6; X = I or Br): Low-Temperature Miscibility, Additivity, and Tunable Stability Journal of Physical Chemistry C 2023
- A. T. J. Nicolson et al. Cu2SiSe3 as a promising solar absorber: harnessing cation dissimilarity to avoid killer antisites Journal of Materials Chemistry A 2023
- Y. W. Woo, Z. Li, Y-K. Jung, J-S. Park, A. Walsh Inhomogeneous Defect Distribution in Mixed-Polytype Metal Halide Perovskites ACS Energy Letters 2023
- P. A. Hyde et al. Lithium Intercalation into the Excitonic Insulator Candidate Ta2NiSe5 Inorganic Chemistry 2023
- J. Willis, K. B. Spooner, D. O. Scanlon On the possibility of p-type doping in barium stannate Applied Physics Letters 2023
- J. Cen et al. Cation disorder dominates the defect chemistry of high-voltage LiMn1.5Ni0.5O4 (LMNO) spinel cathodes Journal of Materials Chemistry A 2023
- J. Willis & R. Claes et al. Limits to Hole Mobility and Doping in Copper Iodide Chemistry of Materials 2023
- I. Mosquera-Lois & S. R. Kavanagh, A. Walsh, D. O. Scanlon Identifying the ground state structures of point defects in solids npj Computational Materials 2023
- Y. T. Huang & S. R. Kavanagh et al. Strong absorption and ultrafast localisation in NaBiS2 nanocrystals with slow charge-carrier recombination Nature Communications 2022
- S. R. Kavanagh, D. O. Scanlon, A. Walsh, C. Freysoldt Impact of metastable defect structures on carrier recombination in solar cells Faraday Discussions 2022
- Y-S. Choi et al. Intrinsic Defects and Their Role in the Phase Transition of Na-Ion Anode Na2Ti3O7 ACS Applied Energy Materials 2022
- S. R. Kavanagh, D. O. Scanlon, A. Walsh Rapid Recombination by Cadmium Vacancies in CdTe ACS Energy Letters 2021
- C. J. Krajewska et al. Enhanced visible light absorption in layered Cs3Bi2Br9 through mixed-valence Sn(II)/Sn(IV) doping Chemical Science 2021
doped (née DefectsWithTheBoys #iykyk) has benefitted from feedback from many users, in particular
members of the Scanlon and Walsh research groups who have used / are using it in their work. Direct contributors are listed in the Contributors sidebar above; including Seán Kavanagh, Alex Squires, Adair Nicolson, Irea Mosquera-Lois, Alex Ganose, Bonan Zhu, Katarina Brlec, Sabrine Hachmioune and Savya Aggarwal.
doped was originally based on the excellent PyCDT (no longer maintained), but transformed and morphed over time as more and more functionality was added. After breaking changes in pymatgen, the package was entirely refactored and rewritten, to work with the new
pymatgen-analysis-defects package.