-
Notifications
You must be signed in to change notification settings - Fork 6
Commit
This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository.
Fixed links in the README and produced a Python-specific README_PYPI …
…for Pypi
- Loading branch information
1 parent
c1bfac2
commit cfd3981
Showing
4 changed files
with
192 additions
and
27 deletions.
There are no files selected for viewing
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,163 @@ | ||
| # PhyloDeep | ||
|
|
||
| PhyloDeep is a python library for parameter estimation and model selection from phylogenetic trees, based on deep learning. | ||
|
|
||
| For more information on the method, including the covered parameter subspace, please refer to the preprint here: [bioRxiv](https://www.biorxiv.org/content/10.1101/2021.03.11.435006v3). | ||
|
|
||
| The installation time of the package can be up to several minutes, including downloading dependencies. The run time | ||
| should be a couple of seconds. The package was tested in Linux (Ubuntu 18.08), Windows 10 and MacOS. | ||
|
|
||
| ## Installation | ||
|
|
||
| ### Windows | ||
| For **Windows** users, we recommend installing __phylodeep__ via [Cygwin environment](https://www.cygwin.com/). | ||
| First instal Python 3.6 and pip3 from the Cygwin packages. Then install __phylodeep__: | ||
| ```bash | ||
| pip3 install phylodeep | ||
| ``` | ||
|
|
||
| ### All other platforms | ||
|
|
||
| You can install __phylodeep__ for Python 3.6 with or without [conda](https://conda.io/docs/), following the procedures described below: | ||
|
|
||
| #### Installing with conda | ||
|
|
||
| Once you have conda installed, create an environment for __phylodeep__ with Python 3.6 (here we name it phyloenv): | ||
|
|
||
| ```bash | ||
| conda create --name phyloenv python=3.6 | ||
| ``` | ||
|
|
||
| Then activate it: | ||
| ```bash | ||
| conda activate phyloenv | ||
| ``` | ||
|
|
||
| Then install __phylodeep__ in it: | ||
|
|
||
| ```bash | ||
| pip install phylodeep | ||
| ``` | ||
|
|
||
| #### Installing without conda | ||
|
|
||
| Make sure that Pyhon 3.6 and pip3 are installed, then install __phylodeep__: | ||
|
|
||
| ```bash | ||
| pip3 install phylodeep | ||
| ``` | ||
|
|
||
| ## Usage | ||
|
|
||
| If you installed __phylodeep__ with conda, do not forget to activate the corresponding environment (e.g. phyloenv) before using PhyloDeep: | ||
| ```bash | ||
| conda activate phyloenv | ||
| ``` | ||
|
|
||
| We recommend to perform a priori model adequacy first to assess whether the input data resembles well the | ||
| simulations on which the neural networks were trained. | ||
|
|
||
| ### Example data | ||
|
|
||
| Here, we use an HIV tree reconstructed from 200 sequences, published in "Phylodynamics on local sexual contact networks" | ||
| by Rasmussen _et al._ [[PLoS Comput. Biol. 2017]](https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1005448), | ||
| which you can find at [PairTree GitHub](https://github.com/davidrasm/PairTree) | ||
| and in [test_tree_HIV_Zurich/Zurich.trees](https://github.com/evolbioinfo/phylodeep/blob/main/test_tree_HIV_Zurich/Zurich.trees). | ||
|
|
||
| ### Python | ||
|
|
||
| ```python | ||
| from phylodeep import BD, BDEI, BDSS, FULL | ||
| from phylodeep.checkdeep import checkdeep | ||
| from phylodeep.modeldeep import modeldeep | ||
| from phylodeep.paramdeep import paramdeep | ||
|
|
||
|
|
||
| path_to_tree = './Zurich.trees' | ||
|
|
||
| # set presumed sampling probability | ||
| sampling_proba = 0.25 | ||
|
|
||
| # a priori check for models BD, BDEI, BDSS | ||
| checkdeep(path_to_tree, model=BD, outputfile_png='BD_a_priori_check.png') | ||
| checkdeep(path_to_tree, model=BDEI, outputfile_png='BDEI_a_priori_check.png') | ||
| checkdeep(path_to_tree, model=BDSS, outputfile_png='BDSS_a_priori_check.png') | ||
|
|
||
|
|
||
| # model selection | ||
| model_BDEI_vs_BD_vs_BDSS = modeldeep(path_to_tree, sampling_proba, vector_representation=FULL) | ||
|
|
||
| # the selected model is BDSS | ||
|
|
||
| # parameter inference | ||
| param_BDSS = paramdeep(path_to_tree, sampling_proba, model=BDSS, vector_representation=FULL, | ||
| ci_computation=True) | ||
|
|
||
| # for the interpretation of results, please see below | ||
| ``` | ||
|
|
||
| ### Command line | ||
|
|
||
| ```bash | ||
|
|
||
| # we use here a tree of 200 tips | ||
|
|
||
| # a priori model adequacy check: highly recommended | ||
| checkdeep -t ./Zurich.trees -m BD -o BD_model_adequacy.png | ||
| checkdeep -t ./Zurich.trees -m BDEI -o BDEI_model_adequacy.png | ||
| checkdeep -t ./Zurich.trees -m BDSS -o BDSS_model_adequacy.png | ||
|
|
||
| # model selection | ||
| modeldeep -t ./Zurich.trees -p 0.25 -v CNN_FULL_TREE -o model_selection.csv | ||
|
|
||
| # parameter inference | ||
| paramdeep -t ./Zurich.trees -p 0.25 -m BDSS -v CNN_FULL_TREE -o HIV_Zurich_BDSS_CNN.csv | ||
| paramdeep -t ./Zurich.trees -p 0.25 -m BDSS -v FFNN_SUMSTATS -o HIV_Zurich_BDSS_FFNN_CI.csv -c | ||
| ``` | ||
|
|
||
| ### Example of output and interpretations | ||
|
|
||
| The a priori model adequacy check results in the following figures: | ||
|
|
||
| #### BD model adequacy test | ||
|  | ||
|
|
||
| #### BDEI model adequacy test | ||
|  | ||
|
|
||
| #### BDSS model adequacy test | ||
| 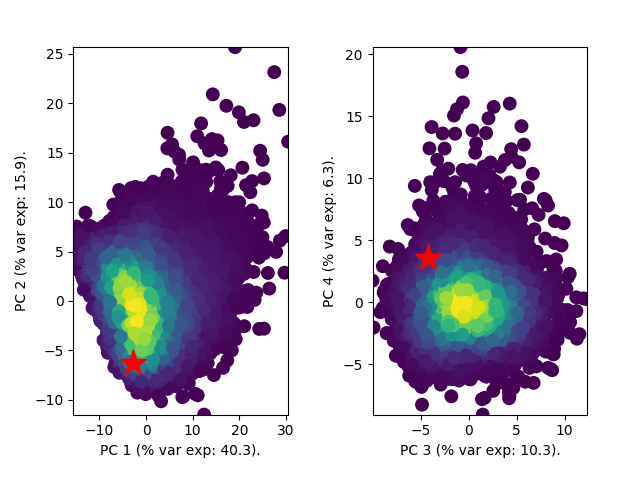 | ||
|
|
||
| For the three models (BD, BDEI and BDSS), HIV tree datapoint (represented by a red star) is well inside the data cloud | ||
| of simulations, where warm colors correspond to high density of simulations. The simulations and HIV tree datapoint were | ||
| in the form of summary statistics prior to applying PCA. All three models thus pass the model adequacy check. | ||
|
|
||
| We then apply model selection using the full tree representation and obtain the following result: | ||
|
|
||
| | Model | Probability BDEI | Probability BD | Probability BDSS | | ||
| | -------- | ------------- | ------------- | ------------- | | ||
| | __Predicted probability__ | 0.00 | 0.00 | 1.00 | | ||
|
|
||
| The BDSS probability is by far the highest: it is the BDSS model that is confidently selected | ||
|
|
||
| Finally, under the selected model BDSS, we predict parameter values together with 95% CIs: | ||
|
|
||
| | | R naught | Infectious period | X transmission | Superspreading fraction | | ||
| | ------------- | ------------- | ------------- | ------------- | ------- | | ||
| | __predicted value__ | 1.69 | 9.78 | 9.34 | 0.079 | | ||
| | __CI 2.5%__ | 1.40 | 8.12 | 6.65 | 0.050 | | ||
| | __CI 97.5%__ | 2.08 | 12.26 | 10 | 0.133 | | ||
|
|
||
| The point estimates for parameters that are no time related (R naught, X transmission and Superspreading fraction) are | ||
| well inside the parameter ranges of simulations and thus seem valid (R naught between 1 and 5, x transmission between 3 | ||
| and 10, superspreading fraction between 0.05 and 0.20). | ||
|
|
||
|
|
||
| The time related parameters (infectious and eventually incubation period for BDEI model) are in the same units as the | ||
| branches of input tree, here in years (9.78 years). The covered parameter space for time related parameters is large | ||
| due to internal rescaling of all input trees. It should apply to any tree. | ||
|
|
||
| ## Preprint | ||
|
|
||
| Voznica J, Zhukova A, Boskova V, Saulnier E, Lemoine F, Moslonka-Lefebvre M, Gascuel O (2022) | ||
| __Deep learning from phylogenies to uncover the transmission dynamics of epidemics__. [bioRxiv](https://www.biorxiv.org/content/10.1101/2021.03.11.435006v3) |
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters