Metabuli classifies metagenomic reads by comparing them to reference genomes. You can use Metabuli to profile the taxonomic composition of your samples or to detect specific (pathogenic) species.
Sensitive and Specific. Metabuli uses a novel k-mer structure, called metamer, to analyze both amino acid (AA) and DNA sequences. It leverages AA conservation for sensitive homology detection and DNA mutations for specific differentiation between closely related taxa.
A laptop is enough. Metabuli operates within user-specified RAM limits, allowing it to search any database that fits in storage. A PC with 8 GiB of RAM is sufficient for most analyses.
A few clicks are enough. Metabuli App is now available here. With just a few clicks, you can run Metabuli and browse the results with Sankey and Krona plots on your PC.
Short reads, long reads, and contigs. Metabuli can classify all types of sequences.
For more details, please see Nature Methods, PDF, bioRxiv, or ISMB 2023 talk.

🖥️ Metabuli App for Windows, MacOS, and Linux are now available!
Run taxonomic profiling in just a few clicks and explore results with Sankey and Krona plots.
Download the app for your OS here—no separate Metabuli installation needed.
- Added
extractmodule to extract reads classified into a certain taxon.
- Metabuli became faster 🚀
- Windows: 8.3 times faster
- MacOS: 1.7 times faster
- Linux: 1.3 times faster
- Test details are in release note.
- Fixed a bug in score calculation that could affect classification results.
- Windows OS is supported.
Metabuli v1.0.6 is too slow on Windows OS. Please use v1.0.7 or later.
- Fixed a minor reproducibility issue.
- Fixed a performance-harming bug occurring with sequences containing lowercased bases.
- Auto adjustment of
--match-per-kmerparameter. Issue #20 solved. - Record version info. in
db.parameter
# install via conda
conda install -c conda-forge -c bioconda metabuli
# Linux AVX2 build (fast, recommended for most Linux system
# check using: cat /proc/cpuinfo | grep avx2)
wget https://mmseqs.com/metabuli/metabuli-linux-avx2.tar.gz; tar xvzf metabuli-linux-avx2.tar.gz; export PATH=$(pwd)/metabuli/bin/:$PATH
# Linux SSE2 build (slower, for old systems)
wget https://mmseqs.com/metabuli/metabuli-linux-sse2.tar.gz; tar xvzf metabuli-linux-sse2.tar.gz; export PATH=$(pwd)/metabuli/bin/:$PATH
# MacOS (Universal, works on Apple Silicon and Intel Macs)
wget https://mmseqs.com/metabuli/metabuli-osx-universal.tar.gz; tar xvzf metabuli-osx-universal.tar.gz; export PATH=$(pwd)/metabuli/bin/:$PATH
Metabuli also works on Linux ARM64 and Windows systems. Please check https://mmseqs.com/metabuli for static builds for other architectures.
To compile Metabuli from source code use the following commands:
git clone https://github.com/steineggerlab/Metabuli.git
cd Metabuli
mkdir build && cd build
cmake -DCMAKE_BUILD_TYPE=Release ..
make -j 16
The built binary can be found in ./build/src.
You can download pre-built databases using databases workflow.
NOTE: The databases workflow may not work if you don't use the latest version of Metabuli.
In that case, please manually download databases from this link.
Usage:
metabuli databases DB_NAME OUTDIR tmp
# NOTE
- A human genome (T2T-CHM13v2.0) is included in all databases below.
1. RefSeq Virus (8.1 GiB)
- NCBI RefSeq release 223 virus genomes
- Database will be in OUT_DIR/refseq_virus
metabuli databases RefSeq_virus OUT_DIR tmp
2. RefSeq Prokaryote and Virus (115.6 GiB)
- RefSeq prokaryote genomes (Complete Genome/Chromosome, 2024-03-26) + RefSeq Virus above.
- Database will be in OUT_DIR/refseq_prokaryote_virus
metabuli databases RefSeq OUTDIR tmp
3. GTDB (101 GiB)
- GTDB 214.1 (Complete Genome/Chromosome, CheckM completeness > 90 and contamination < 5).
- Database will be in OUT_DIR/gtdb
metabuli databases GTDB OUTDIR tmp
4. RefSeq Releases 224 (619 GiB)
- Viral and prokaryotic genomes of RefSeq release 224.
metabuli databases RefSeq_release OUTDIR tmp
Downloaded files are stored in OUTDIR/DB_NAME directory, which can be provided for classify module as DBDIR.
metabuli classify <i:FASTA/Q> <i:DBDIR> <o:OUTDIR> <Job ID> [options]
- INPUT : FASTA/Q file of reads you want to classify. (gzip supported)
- DBDIR : The directory of reference DB.
- OUTDIR : The directory where the result files will be generated.
- Job ID: It will be the prefix of result files.
# Paired-end
metabuli classify read_1.fna read_2.fna dbdir outdir jobid
# Single-end
metabuli classify --seq-mode 1 read.fna dbdir outdir jobid
# Long-read
metabuli classify --seq-mode 3 read.fna dbdir outdir jobid
* Important parameters:
--threads : The number of threads used (all by default)
--max-ram : The maximum RAM usage. (128 GiB by default)
--min-score : The minimum score to be classified
--min-sp-score : The minimum score to be classified at or below species rank.
--taxonomy-path: Directory where the taxonomy dump files are stored. (DBDIR/taxonomy by default)
--accession-level : Set 1 to use accession level classification (0 by default).
It is available when the DB is also built with accession level taxonomy.
- Paratemers for precision mode (Metabuli-P)
- Illumina short reads:
--min-score 0.15 --min-sp-score 0.5 - PacBio HiFi reads:
--min-score 0.07 --min-sp-score 0.3 - PacBio Sequel II reads:
--min-score 0.005 - ONT reads:
--min-score 0.008
- Illumina short reads:
This will generate three result files: JobID_classifications.tsv, JobID_report.tsv, and JobID_krona.html.
Sankey diagram is available in the GUI app.
- Classified or not
- Read ID
- Taxonomy identifier
- Effective read length
- DNA level identity score
- Classification Rank
- List of "taxID : k-mer match count"
1 read_1 2688 294 0.627551 subspecies 2688:65
1 read_2 2688 294 0.816327 subspecies 2688:78
0 read_3 0 294 0 no rank
The proportion of reads that are assigned to each taxon.
33.73 77571 77571 0 no rank unclassified
66.27 152429 132 1 no rank root
64.05 147319 2021 8034 superkingdom d__Bacteria
22.22 51102 3 22784 phylum p__Firmicutes
22.07 50752 361 22785 class c__Bacilli
17.12 39382 57 123658 order o__Bacillales
15.81 36359 3 126766 family f__Bacillaceae
15.79 36312 26613 126767 genus g__Bacillus
2.47 5677 4115 170517 species s__Bacillus amyloliquefaciens
0.38 883 883 170531 subspecies RS_GCF_001705195.1
0.16 360 360 170523 subspecies RS_GCF_003868675.1
0.11 248 248 170525 subspecies RS_GCF_002209305.1
0.02 42 42 170529 subspecies RS_GCF_002173635.1
0.01 24 24 170539 subspecies RS_GCF_000204275.1
It is for an interactive taxonomy report (Krona). You can use any modern web browser to open JobID_krona.html.
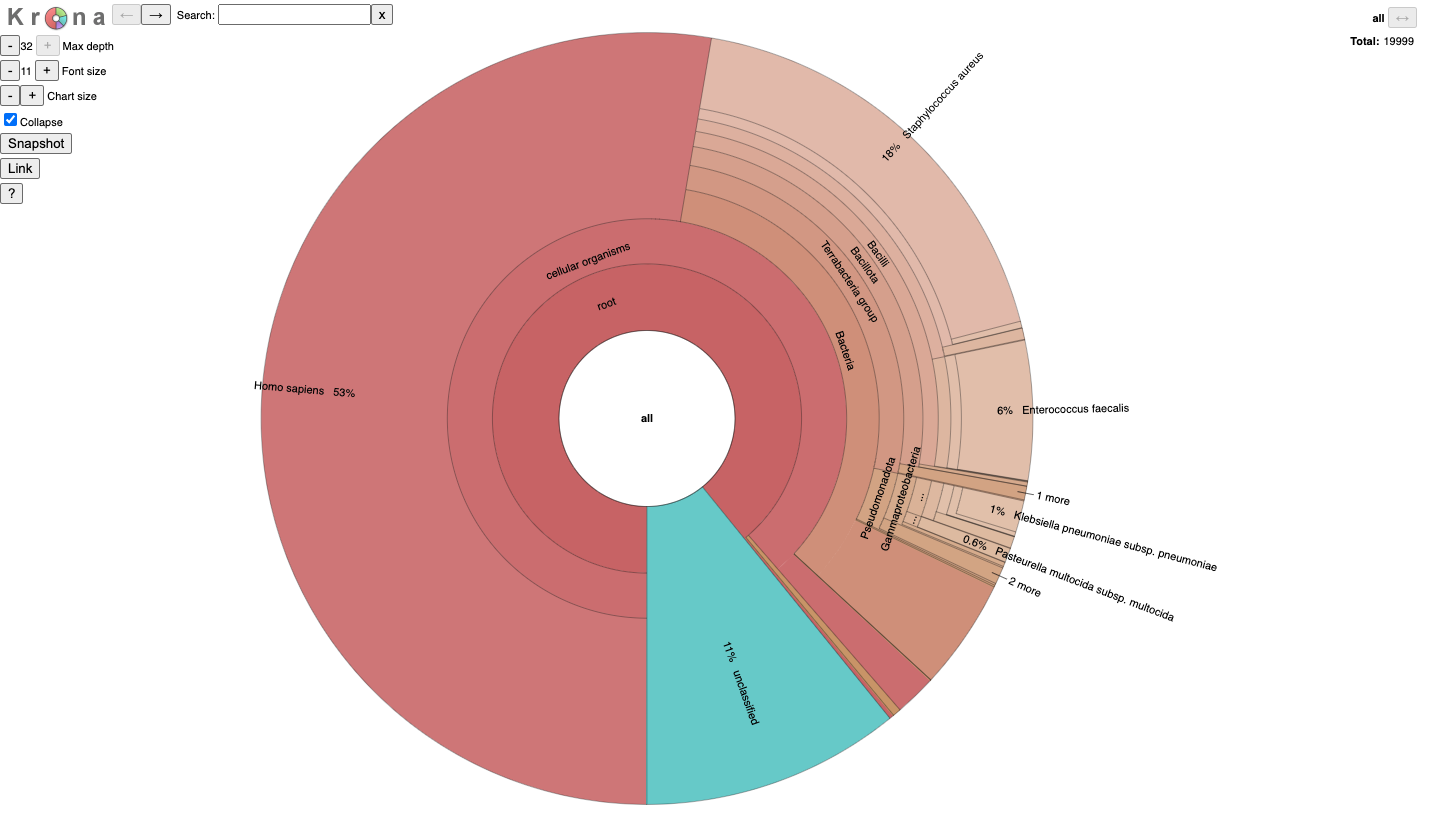
Metabuli can classify reads against a database of any size as long as the database is fits in the hard disk, regardless of the machine's RAM size. We tested it with a MacBook Air (2020, M1, 8 GiB), where we classified about 15 M paired-end 150 bp reads (~5 GiB in size) against a database built with ~23K prokaryotic genomes (~69 GiB in size).
After running the classify command, you can extract reads that are classified under a specific taxon.
This requires the FASTA/Q files used in the classify step and the JobID_classifications.tsv file, which is generated as one of the output files.
metabuli extract <i:FASTA/Q> <i:read-by-read classification> <i:DBDIR> --tax-id TAX_ID
- FASTA/Q : The FASTA/Q file(s) used during the `classify` step.
- read-by-read classification : The JobID_classifications.tsv file generated by the `classify` step.
- DBDIR : The same DBDIR used in the `classify` step.
- TAX_ID : The taxonomy ID of the taxon at any rank (e.g., species, genus) from which you want to extract the reads.
# Paired-end
metabuli extract read_1.fna read_2.fna JobID_classifications.tsv dbdir --tax-id TAX_ID
# Single-end
metabuli extract --seq-mode 1 read.fna JobID_classifications.tsv dbdir --tax-id TAX_ID
# Long-read
metabuli extract --seq-mode 3 read.fna JobID_classifications.tsv dbdir --tax-id TAX_ID
- For paired-end samples:
read_1_TAX_ID.fnaandread_2_TAX_ID.fna - For single-end or long-read samples:
read_TAX_ID.fna
To build a custom database, you need three things:
- FASTA files : Each sequence of your FASTA files must be separated by '>accession.version' like '>CP001849.1'. The accession doesn't have to follow the NCBI format, but it must be unique and included in the accession2taxid file.
- accession2taxid : Mapping from accession to taxonomy ID. The sequences whose accessions are not listed here will be skipped.
- NCBI-style taxonomy dump : 'names.dmp' , 'nodes.dmp', and 'merged.dmp' are required. The sequences whose taxonomy IDs are not included here will be skipped.
The steps for building a database with NCBI or GTDB taxonomy are described below.
- If you want to include a custom sequence, edit
accession2taxidandtaxdumpfiles properly as follows.accession2taxid- For a sequence whose header is
>custom, addcustom[tab]custom[tab]taxid[tab]anynumber. - As above, version number is not necessary.
taxidmust be included in thenodes.dmpandnames.dmp.- Put any number for the last column. It is not used in Metabuli.
- For a sequence whose header is
taxdump- Edit
nodes.dmpandnames.dmpif you introduced a newtaxidinaccession2taxid.
- Edit
metabuli add-to-library <FASTA list> <accession2taxid> <DBDIR>
- FASTA list : A file containing absolute paths of each FASTA file.
- accession2taxid : A path to NCBI-style accession2taxid.
- DBDIR : Sequences will be stored in 'DBDIR/library'.
* Option
--taxonomy-path: Directory of taxdump files. (DBDIR/taxonomy by default)
* NOTE: When resume is needed, remove the files in DBDIR/library and run the command again.
It groups your sequences into separate files according to their species.
Accessions that are not included in the <accession2taxid> will be skipped and listed in unmapped.txt.
# Get the list of absoulte paths of files in your library
find <DBDIR>/library -type f -name '*.fna' > library-files.txt
metabuli build <DBDIR> <LIB_FILES> <accession2taxid> [options]
- DBDIR: The same DBDIR from the previous step.
- LIB_FILES: A file containing absolute paths of the FASTA files in DBDIR/library (library-files.txt)
- accession2taxid : A path to NCBI-style accession2taxid.
* Options
--threads : The number of threads used (all by default)
--max-ram : The maximum RAM usage. (128 GiB by default)
--taxonomy-path : Directory where the taxonomy dump files are stored. (DBDIR/taxonomy by default)
--accession-level : Set 1 to creat a DB for accession level classification (0 by default).
This will generate diffIdx, info, split, and taxID_list and some other files. You can delete '*_diffIdx' and '*_info' if generated.
Requirements:
You need assembly FASTA files whose file name (or path) includes the assembly accession.
If you downloaded assemblies using ncbi-genome-download, you probably don't have to care about it.
The regular expression of assembly accessions is (GC[AF]_[0-9].[0-9])
# 1.
# 1-1. Move to the 'util' directory
cd METABULI_DIR/util
# 1-2. Run prepare_gtdb_taxonomy.sh
./prepare_gtdb_taxonomy.sh <DBDIR>
- DBDIR : Result files are stored in 'DBDIR/taxonomy'.
** Please clone Metabuli's repository to use this script.
** It is not provided in the precompiled binaries or bioconda package.
In DBDIR/taxonomy, it will generate taxonomy dmp files and assacc_to_taxid.tsv with other files.
# 2.
metabuli add-to-library <FASTA list> <accession2taxid> <DBDIR> --assembly true
- FASTA list : A file containing absolute paths of each assembly file.
Each path must include a corresponding assembly accession.
- accession2taxid : 'assacc_to_taxid.tsv' from the previous step
- DBDIR : The same DBDIR from the previous step.
** When resume is needed, remove the files in DBDIR/library and run the command again.
This will add your FASTA files to DBDIR/library according to their species taxonomy ID and generate 'my.accession2taxid'
# Get the list of absoulte paths of files in your library
find <DBDIR>/library -type f -name '*.fna' > library-files.txt
metabuli build <DBDIR> <LIB_FILES> <accession2taxid> [options]
- DBDIR: The same DBDIR from the previous step.
- <LIB_FILES>: A file containing absolute paths of the FASTA files in DBDIR/library (library-files.txt)
- accession2taxid : A path to NCBI-style accession2taxid.
* Options
--threads : The number of CPU-cores used (all by default)
--taxonomy-path: Directory where the taxonomy dump files are stored. (DBDIR/taxonomy by default)
--reduced-aa : 0. Use 20 alphabets or 1. Use 15 alphabets to encode amino acids.
--spacing-mask : Binary mask for spaced metamer. The same mask must be used for DB creation and classification.
A mask should contain at least eight '1's, and '0' means skip.
--accession-level : Set 1 to use accession level taxonomy (0 by default).
This will generate diffIdx, info, split, and taxID_list and some other files. You can delete '*_diffIdx' and '*_info' if generated.
You can add new sequences to an existing database. The taxonomy information you provide here must be compatible with the existing database.
# 1. Add new sequences to the library
metabuli add-to-library <FASTA list> <accession2taxid> <DBDIR>
- FASTA list : A file of absolute paths to FASTA files.
- accession2taxid : A path to NCBI-style accession2taxid.
- DBDIR : Sequences will be stored in 'DBDIR/library'.
* Option
--taxonomy-path: Directory of taxonomy dump files. (DBDIR/taxonomy by default)
# 2. Get the list of absoulte paths of files in your library
find <DBDIR>/library -type f -name '*.fna' > library-files.txt
# 3. Add new sequences to the existing database
metabuli <new DB directory> <FASTA list> <accesssion2taxid> <old DB directory>
- FASTA list: A file containing absolute paths of the FASTA files in DBDIR/library (library-files.txt)
- accession2taxid : A path to NCBI-style accession2taxid.
* Options
--threads : The number of threads used (all by default)
--max-ram : The maximum RAM usage. (128 GiB by default)
--taxonomy-path : Directory of taxonomy dump files. (DBDIR/taxonomy by default)
--accession-level : Set 1 to creat a DB for accession level classification (0 by default).
The example here was detecting SARS-CoV-2 variant-specific reads, but has changed since the pre-built DB no longer contains the variant genomes.
Classifying RNA-seq reads from a COVID-19 patient. The whole process must take less than 10 mins using a personal machine.
metabuli databases RefSeq_virus OUTDIR tmp
fasterq-dump --split-files SRR14484345
Download SRA Toolkit containing
fasterq-dumphere
metabuli classify SRR14484345_1.fq SRR14484345_2.fq OUTDIR/refseq_virus RESULT_DIR JOB_ID --max-ram RAM_SIZE
Find a section like the example below
92.2331 510490 442 species 694009 Severe acute respiratory syndrome-related coronavirus
92.1433 509993 509993 no rank 2697049 Severe acute respiratory syndrome coronavirus 2