![]()
For full documentation, refer to the package documentation site.
The goal of bedtorch is to provide a fast BED file manipulation tool suite native in R.
In bioinformatics studies, an important type of jobs is related to BED and BED-like file manipulation. To name a few example:
- Filtering cfDNA fragments based on length
- Identifying genomic regions overlapping with certain genes
- Filtering features based on epigenetic profiles such as histone modification levels
- Extracting SNVs in a certain genomic interval
Many of such tasks can be done by some highly-optimized tools, such as bedtools and bedops. However, these are binary tools which only works in shell. Sometimes, especially for developers using R, it’s desirable to invoke these tools in R codes.
One solution is using functions that invokes system commands
(system2
or
system).
bedr,
a popular package, is one good example. It’s a bridge linking between R
codes and shell tools. Under the hood, it writes data into the temporary
directory, calls certain command line tools such as tabix, bedtools,
or bedops, parses the output and re-generate a data frame in R space.
Generally this approach is very helpful. However, it’s worth noting some disadvantages.
- This workflow requires lots of disk IO, since data frames in R needs to be written to temporary directories before being fed to command line tools.
- Command line output is captured and transferred back to R s space.
The data transfer is done by stdin/stdout pipes. For R, reading
large amount of text content from stdin can be extremely slow. As a
result, most of time when large dataset is involved,
bedris hardly usable. bedrcaptures command line output as plain text. Therefore, when the data is transferred back to R, most of the information about data types, index (in the case ofdata.table), column names, etc., are lost. It’s the package user’s responsibility to make sure data types are correct, column names are restored, column indices have been rebuilt (in the case ofdata.table, which can be very time consuming). Often this job is tedious, error-prone and introduces very large overheads.- It requires the command line tools to be present in
PATH. This depends on the end user’s system environment and the R developer can do nothing about it. It puts additional burden to deployment, especially when the end user is not an IT professional.
Another approach is using native R data types to represent BED-like
data. R’s native data.frame seems to be a good model, but in many
scenarios, it’s performance is poor. More importantly, it lacks many
advanced features compared with GenomicRanges, dplyr and
data.table.
GenomicRanges is widely used in bioinformatics community. Although it
provides essential building blocks for many tasks, such as
findOverlaps, reduce, etc., it doesn’t provide high-level features
as bedtools does. Using findOverlaps, reduce, and other functions
provided by GenomicRanges, users may implement operations such as map,
merge, unique intersect, but this still requires lots of coding.
It would be desirable to have an R package, which represent BED-like
data by GenomicRanges or data.table objects, and provide native
support for common BED manipulations, such as merge, intersect, filter,
etc.
bedtorch is an attempt for this. Under the hood, every BED dataset is
represented as a GenomicRanges object (it also can represent the
dataset as data.table, if preferred. But this is not recommended).
Users can apply various operations on it, such as intersect, merge, etc.
In terms of the core computation, bedtorch’s performance is comparable
to bedtools. However, since no disk IO and data conversion is needed,
users can directly manipulate the dataset in memory, therefore in
practice, via bedtorch, many tasks can be done about one magnitude
faster.
Additionally, by linking to
htslib, bedtorch can write data
frames directly to BGZIP-format files, and optionally create the tabix
index. It can also directly load either local or remote BGZIP-format
files at any particular genomic regions. This feature is done by htslib,
therefore not requiring external command line tools such as tabix or
bgzip. In the case of working with remote files, being capable of
loading any arbitrary portion of the file will be very convenient.
BGZIP is a variant of gzip and
is widely used in bioinformatics. It’s compatible with gzip, with an
important additional feature: allows a compressed file to be indexed for
fast query. However, R is unaware of BGZIP. It considers all .gz files
as gzip-compressed, thus doesn’t take advantage of BED index, and cannot
output BGZIP files.
For more details, refer to the package reference page.
# install.packages("devtools")
devtools::install_github("haizi-zh/bedtorch")Here are several basic examples which shows you how to solve a common problem:
Read a BED file from disk:
library(bedtorch)
## Load BED data
file_path <-
system.file("extdata", "example2.bed.gz", package = "bedtorch")
bedtbl <- read_bed(file_path, range = "1:3001-4000")
bedtbl
#> GRanges object with 14 ranges and 2 metadata columns:
#> seqnames ranges strand | score1 score2
#> <Rle> <IRanges> <Rle> | <integer> <integer>
#> [1] 1 2926-3011 * | 106 181
#> [2] 1 3004-3092 * | 88 193
#> [3] 1 3092-3193 * | 118 212
#> [4] 1 3165-3248 * | 94 211
#> [5] 1 3233-3345 * | 107 205
#> ... ... ... ... . ... ...
#> [10] 1 3619-3726 * | 108 172
#> [11] 1 3695-3803 * | 117 214
#> [12] 1 3777-3867 * | 109 195
#> [13] 1 3845-3939 * | 92 199
#> [14] 1 3925-4016 * | 93 197
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengthsRead a remote BGZIP BED file for a certain region:
# Load remote BGZIP files with tabix index specified
# Here we need to explicitly indicate `compression` as `bgzip` since we are
# using short URL for `tabix_index`, so that the function cannot guess
# compression type using the URL
read_bed(
"https://git.io/JYATB",
range = "22:20000001-30000001",
tabix_index = "https://git.io/JYAkT",
compression = "bgzip"
)
#> GRanges object with 21 ranges and 2 metadata columns:
#> seqnames ranges strand | score comp_method
#> <Rle> <IRanges> <Rle> | <numeric> <character>
#> [1] 22 20000001-20500000 * | -0.0613535 cofrag/3
#> [2] 22 20500001-21000000 * | -0.0422752 cofrag/3
#> [3] 22 21000001-21500000 * | 0.0268175 cofrag/3
#> [4] 22 21500001-22000000 * | 0.0386308 cofrag/3
#> [5] 22 22000001-22500000 * | 0.0265141 cofrag/3
#> ... ... ... ... . ... ...
#> [17] 22 28000001-28500000 * | 0.08472735 cofrag/3
#> [18] 22 28500001-29000000 * | 0.15800267 cofrag/3
#> [19] 22 29000001-29500000 * | 0.14646452 cofrag/3
#> [20] 22 29500001-30000000 * | 0.07898761 cofrag/3
#> [21] 22 30000001-30500000 * | 0.00434186 cofrag/3
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengthsWrite the previous data table to the temporary directory, and create the tabix index alongside:
write_bed(bedtbl,
file_path = tempfile(fileext = ".bed.gz"),
tabix_index = TRUE)Merge intervals, and take the mean of score in each merged group:
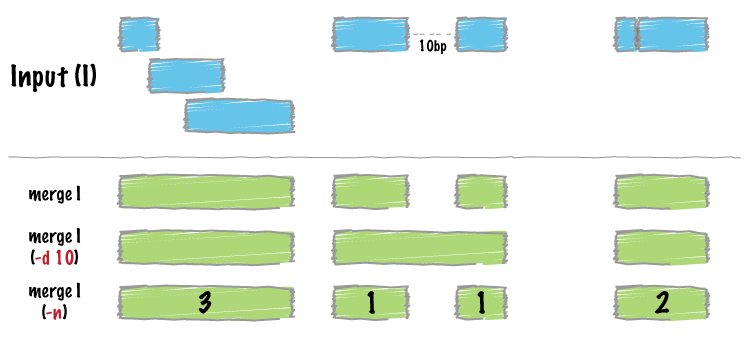
Image by bedtools
operation = list(mean_score = list(on = "score1", func = mean))
merged <- merge_bed(bedtbl, operation = operation)
head(merged)
#> GRanges object with 1 range and 1 metadata column:
#> seqnames ranges strand | mean_score
#> <Rle> <IRanges> <Rle> | <numeric>
#> [1] 1 2926-4016 * | 99.0714
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengthsFind intersections between two datasets:
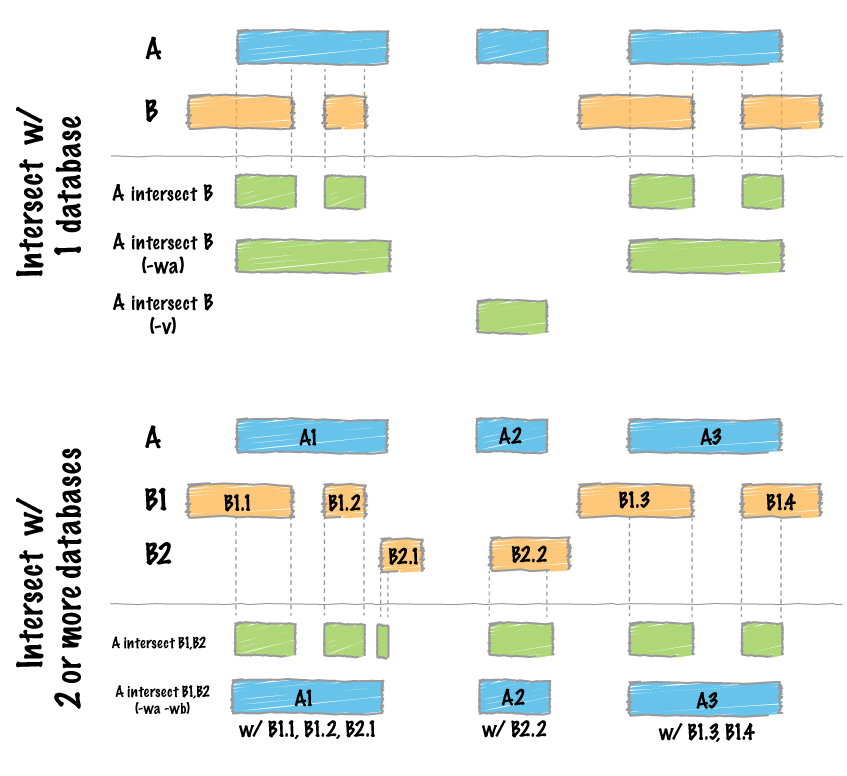
Image by bedtools
file_path1 <- system.file("extdata", "example_merge.bed", package = "bedtorch")
file_path2 <- system.file("extdata", "example_intersect_y.bed", package = "bedtorch")
tbl_x <- read_bed(file_path1, genome = "hs37-1kg")
tbl_y <- read_bed(file_path2, genome = "hs37-1kg")
intersect_bed(tbl_x, tbl_y)
#> GRanges object with 12 ranges and 1 metadata column:
#> seqnames ranges strand | score
#> <Rle> <IRanges> <Rle> | <integer>
#> [1] 21 23-25 * | 7
#> [2] 21 27-30 * | 7
#> [3] 21 30-35 * | 9
#> [4] 21 48-49 * | 1
#> [5] 21 48-50 * | 2
#> ... ... ... ... . ...
#> [8] 22 43-44 * | 9
#> [9] 22 47-50 * | 0
#> [10] 22 52-59 * | 9
#> [11] 22 54-57 * | 5
#> [12] 22 58-60 * | 9
#> -------
#> seqinfo: 84 sequences from hs37-1kg genomeShuffle a BED data table across the genome:
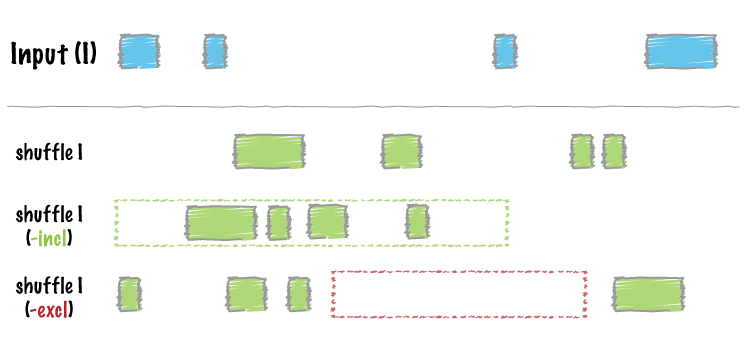
Image by bedtools
shuffle_bed(tbl_x)
#> GRanges object with 20 ranges and 1 metadata column:
#> seqnames ranges strand | score
#> <Rle> <IRanges> <Rle> | <integer>
#> [1] 21 1017987-1017992 * | 9
#> [2] 21 6511120-6511122 * | 5
#> [3] 21 10654657-10654661 * | 4
#> [4] 21 12523054-12523056 * | 7
#> [5] 21 15435384-15435388 * | 2
#> ... ... ... ... . ...
#> [16] 22 30737152-30737159 * | 9
#> [17] 22 31398105-31398108 * | 5
#> [18] 22 33878179-33878183 * | 8
#> [19] 22 35584543-35584546 * | 0
#> [20] 22 44203020-44203028 * | 3
#> -------
#> seqinfo: 84 sequences from hs37-1kg genomeCalculate Jaccard statistics between the two BED data tables:
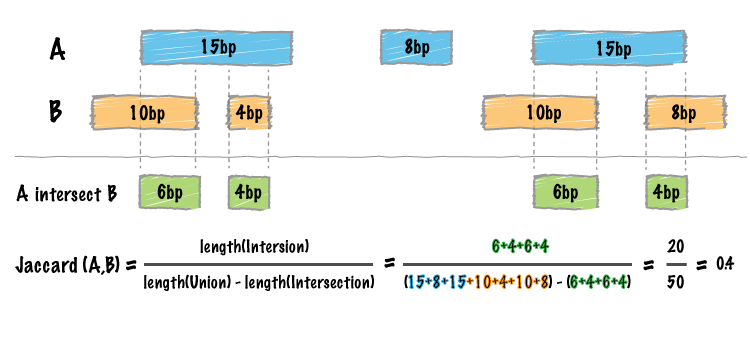
Image by bedtools
jaccard_bed(tbl_x, tbl_y)
#> intersection union jaccard n_intersections
#> 1: 33 94 0.3510638 8The following is the result of a simple benchmark for three common BED manipulations: merge, intersect and map.
Benchmark platform information:
- OS: macOS Big Sur 11.1
- CPU: 2.7 GHz Quad-Core Intel Core i7
- Memory: 16 GB LPDDR3
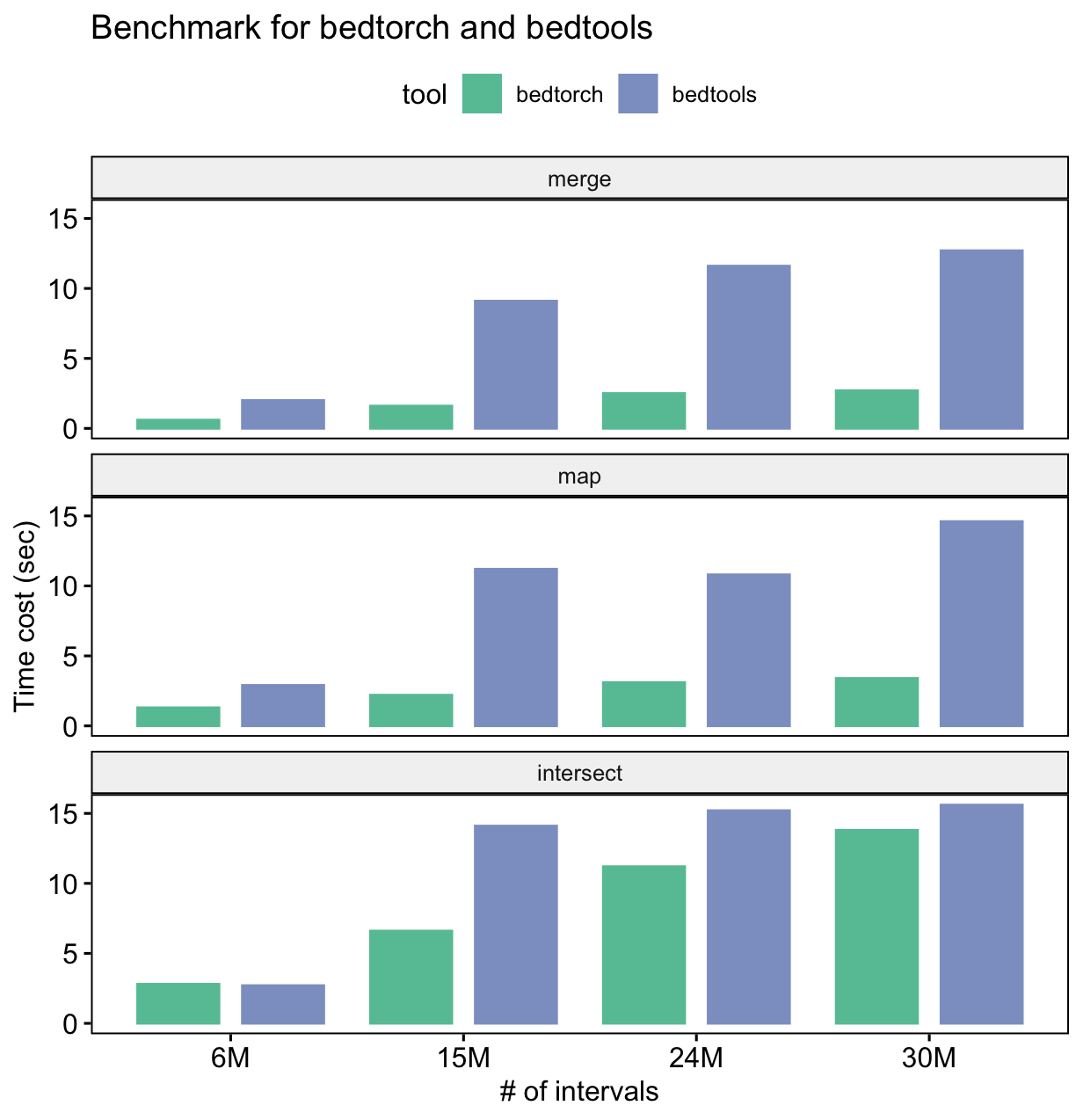
From the benchmark result, we can see that for all three tasks,
bedtorch uses much less time than bedtools.
Note: this does not mean bedtools is indeed slower than bedtorch at
performing the actual computation. Don’t forget bedtools requires
large amount of disk IO, while bedtorch does not.
Many features are inspired by bedtools. Thus, it’s helpful to get familiar with bedtool’s documentation: https://bedtools.readthedocs.io/en/latest/index.html