(m)PTP Tutorial
We will use a dataset of sequences for the lizard genus Gallotia, an endemic genus to Canary islands.
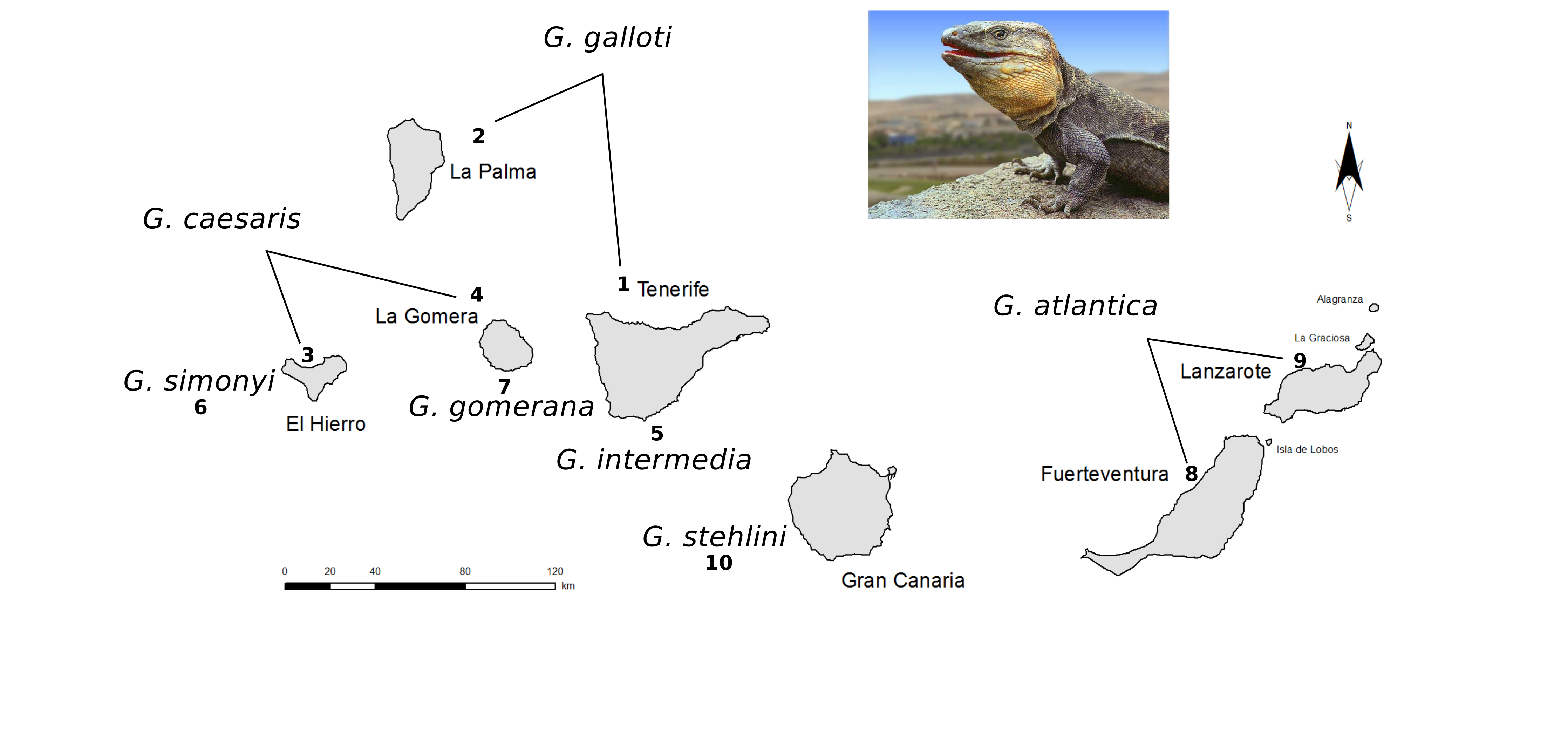
The dataset was originally published by Cox et al., 2010 and it includes three mitochondrial genes COI, 12S and 16S, that have been sequenced for 65 samples. The samples correspond to 7 Gallotia species and 3 outgroup species (Psammodromus algirus, Psammodromus blanci and Lacerta lepida).
The alignment can be found here and the partitioning of the sequences here
We create a folder in our computer and store the alignment and partition file in there
mkdir mptp_demo
cd mptp_demo
wget https://raw.githubusercontent.com/Pas-Kapli/assets/master/mptp_demo/Gallotia_align.fasta
wget https://raw.githubusercontent.com/Pas-Kapli/assets/master/mptp_demo/partitions.txt
We can now infer the tree that we will use as input for (m)PTP. We need a rooted phylogeny for (m)PTP, and we can already do that with raxml:
raxml-ng -model partitions.txt -msa Gallotia_align.fasta --outgroup Lacerta_lepida
we can visualise our tree with a treeviewer (e.g. seaview)
seaview Gallotia_align.fasta.raxml.bestTree
we create a folder for the output of raxml and move everything in there:
mkdir RAxML
mv *raxml* RAxML
First, we create a folder and then we identify the "minbr"
mkdir mptp
cd mptp
mptp --minbr_auto ../Gallotia_align.fasta --tree_file ../RAxML/Gallotia_align.fasta.raxml.bestTree --output_file minbr
The output of this command will look like the following:
mptp 0.2.4_linux_x86_64, 15GB RAM, 8 cores
https://github.com/Pas-Kapli/mptp
Parsing tree file...
Loaded rooted tree...
Parsing FASTA file ../Gallotia_align.fasta...
Computing all pairwise p-distances ...
Minimum branch length (--minbr) should be set to 0.0005270000
We copy the minbr value
mptp --ml --multi --minbr 0.0005270000 --tree_file ../RAxML/Gallotia_align.fasta.raxml.bestTree --output_file multi
This will return two output files: 1) a text file with the command and the delimited cluster multi.txt, and 2) an svg file (multi.svg) that displays the delimitation on a tree.
{kind=link}
Besides the ML delimitation, we also want to know the statistical significance for that result. In this context we run two independent MCMC samplings (--mcmc_runs 2) as follows:
mptp --mcmc 1000000 --mcmc_sample 1000 --mcmc_log 1000 --mcmc_runs 2 --multi --minbr 0.0005270000 --tree_file ../RAxML/Gallotia_align.fasta.raxml.bestTree --output_file MCMC_multi
At the bottom of the screen we will see this:
Preparing overall log-likelihood landscape ...
Overall log-likelihood visualization in MCMC_multi.1628014904.logl.svg ...
Number of edges greater than minimum branch length: 97 / 128
Score Null Model: 288.688182
Best score for multi coalescent rate: 433.009373
Writing delimitation file MCMC_multi.txt ...
Number of delimited species: 11
ML average support based on run with seed 249139162 : 0.92526825665315005
ML average support based on run with seed 1621067196 : 0.92528495087590468
Average standard deviation of support values among runs: 0.000037
Creating tree with combined support values in MCMC_multi.1628014904.combined.tree ...
Creating SVG delimitation file MCMC_multi.1628014904.svg ...
Done...
Both MCMC runs strongly support the ML delimitation (>92%) (how this is calculated?). And they are very similar to each other (Standard deviation of support values among runs 0.000037).
Several files are saved in our working directory (for each of the independent runs and for their combined results). Among them it is worth inspecting the svg file with the combined support values for the delimited clusters MCMC_multi.1628014904.combined.svg and the combined trace file with the sampled likelihood values log file for identifying convergence problems (see here for what you might observe).
{kind=link}
{kind=link}
We get out of the mptp directory and create a new one for the single threshold delimitation and then we run the delimitation:
cd ../
cd ptp
mptp --ml --single --minbr 0.0005270000 --tree_file ../RAxML/Gallotia_align.fasta.raxml.bestTree --output_file single
As before we will get two output files: 1) a text file with the command and the delimited clusters single.txt, and 2) an svg file (single.svg) that displays the delimitation on a tree.
{kind=link}
We also estimate the support values with two MCMC runs as before:
mptp --mcmc 1000000 --mcmc_sample 1000 --mcmc_log 1000 --mcmc_runs 2 --single --minbr 0.0005270000 --tree_file ../RAxML/Gallotia_align.fasta.raxml.bestTree --output_file MCMC_single
Again several files are created among which we can inspect the support values MCMC_single.1628014932.combined.svg and the combined trace file with the sampled likelihood values log file for identifying convergence problems.
{kind=link}
{kind=link}