![]()



![]()
doped is a python package for
managing solid-state defect calculations, with functionality to
generate defect structures and relevant competing phases (for chemical potentials), interface with
ShakeNBreak for
defect structure-searching, write VASP input files for defect
supercell calculations, and automatically parse and analyse the results.
Tutorials showing the code functionality and usage are provided on the docs site.
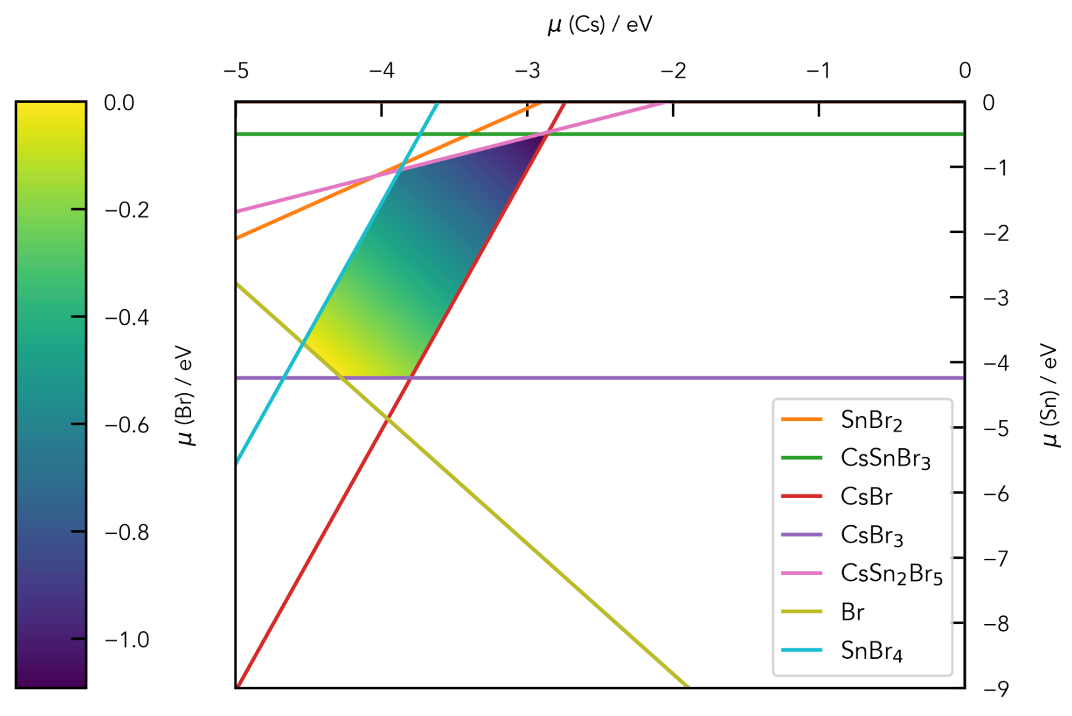
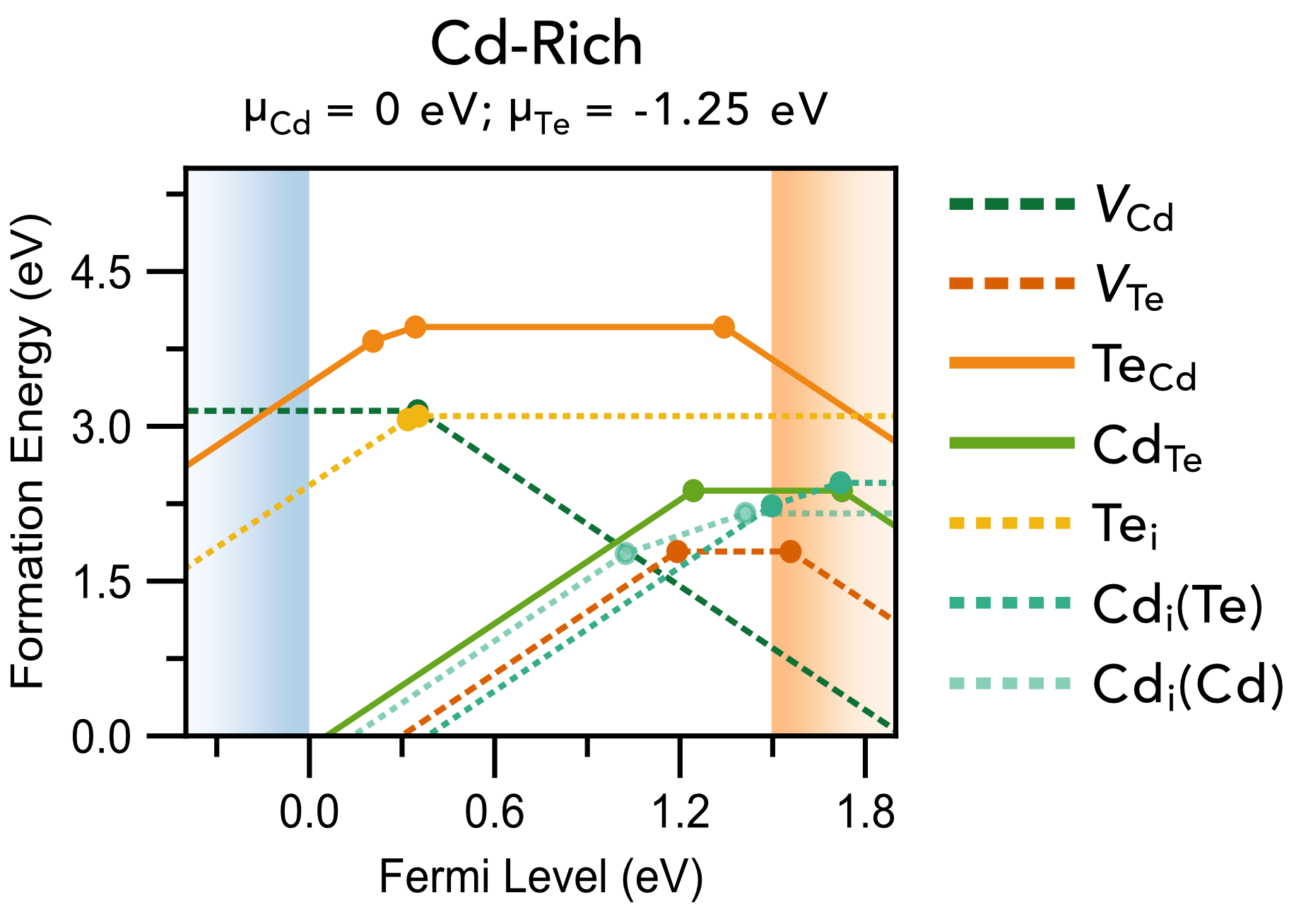
Chemical potential/stability region plots and defect formation energy (a.k.a. transition level) diagrams:
pip install doped # install doped and dependenciesAlternatively if desired, doped can also be installed from conda with:
conda install -c conda-forge dopedIf you haven't done so already, you will need to set up your VASP POTCAR files and Materials Project API with pymatgen using the .pmgrc.yaml file, in order for doped to automatically generate VASP input files for defect calculations and determine competing phases for chemical potentials.
See the docs Installation page for details on this.
As shown in the example notebook, it is highly recommended to use the ShakeNBreak approach when calculating point defects in solids, to ensure you have identified the groundstate structures of your defects. As detailed in the theory paper, skipping this step can result in drastically incorrect formation energies, transition levels, carrier capture (basically any property associated with defects). This approach is followed in the doped example notebook, with a more in-depth explanation and tutorial given on the ShakeNBreak website.
Summary GIF:

SnB CLI Usage:

doped (née DefectsWithTheBoys #iykyk) has benefitted from feedback from many users, in particular
members of the Scanlon and Walsh research groups who have used / are using it in their work. Direct contributors are listed in the Contributors sidebar above; including Seán Kavanagh, Bonan Zhu, Katarina Brlec, Adair Nicolson,
Sabrine Hachmioune and Savya Aggarwal.
Code to efficiently identify defect species from input supercell structures was contributed by Dr
Alex Ganose, and the colour scheme for defect formation energy plots was originally templated from
the aide package, developed by the dynamic duo Adam Jackson and Alex Ganose.
The docs website setup was templated from the ShakeNBreak docs set up by Irea Mosquera-Lois 🙌
doped was originally based on the excellent
PyCDT (no longer maintained), but transformed
and morphed over time as more and more functionality was added. After breaking changes in pymatgen, the package was
entirely refactored and rewritten, to work with the new
pymatgen-analysis-defects package.
- A. T. J. Nicolson et al. Journal of Materials Chemistry A 2023
- Y. W. Woo, Z. Li, Y-K. Jung, J-S. Park, A. Walsh ACS Energy Letters 2023
- P. A. Hyde et al. Inorganic Chemistry 2023
- J. Willis, K. B. Spooner, D. O. Scanlon. ChemRxiv 2023
- X. Wang et al. arXiv 2023
- J. Cen et al. Journal of Materials Chemistry A 2023
- J. Willis & R. Claes et al. ChemRxiv 2023
- I. Mosquera-Lois & S. R. Kavanagh, A. Walsh, D. O. Scanlon npj Computational Materials 2023
- Y. T. Huang & S. R. Kavanagh et al. Nature Communications 2022
- S. R. Kavanagh, D. O. Scanlon, A. Walsh, C. Freysoldt Faraday Discussions 2022
- S. R. Kavanagh, D. O. Scanlon, A. Walsh ACS Energy Letters 2021
- C. J. Krajewska et al. Chemical Science 2021