PRF Model
Original page we are migrating
Dumoulin and Wandell (2008) developed a model-based method that uses the BOLD time course to estimate models of population receptive fields (pRF). Here we describe the mrVista implementation of this method.
- There is a pRF Tutorial page which walks through a particular sample pRF data set.
- There is also a '''Tutorial''' on older "traveling wave" analyses of retinotopy data.
The pRF analysis estimates the parameters of an explicit model of the population of receptive fields within an MR voxel. The simplest pRF model is a two-dimensional circular Gaussian. In addition, we have implemented a difference of circular Gaussian an oval Gaussian model, sum of positive Gaussians, sum of unsigned Gaussians and others. Some of these models are accessible for the graphical interface, but others require evaluation from the command line. We anticipate the list of models will expand over the years.
In the first part of this tutorial, we describe the procedure for fitting the circular, symmetric pRF. For this case the field map is defined by the estimated center positions of the Gaussians, (x,y), and the population receptive field size is determined by the Gaussian standard deviation (sigma). Other pRF summaries, such as the proportion of the receptive field in the contra- and ipsilateral visual field (laterality) can be derived and visualized from the estimated parameters.
Later in the tutorial we explain how to run the code from the command line and write code for more general models. We describe the procedure using the data set which can be downloaded from Not Yet Implemented. This data set measures 3 deg bars moving across visual field, measured and analyzed by Hiroshi Horiguchi. You can follow through this page using the data set.
The general preprocessing steps (motion correction, slice-timing correction, registration to new coordinate frames) are the same as any analysis. The code runs only in the Gray view, so that you must have a segmentation of the subject's brain (see Gray-White Segmentation Pipeline).
The current implementation is built and tested in the "Gray" view. Transform your data into this "Gray" view. (Yes, we are working on the ability to run it from other views).
To transfer your data to the "Gray" view open both "Inplane" and "Gray" window. You can transform the raw data by selecting in the "Gray" window: Xform | Inplane -> Volume | tSeries (all scans) | trilinear interpolation.
An advantage of using the "Gray" view is that you can easily add other fMRI runs from different sessions or days. The fMRI data from different session can be ported into the current fMRI session by selecting in the "Gray" window: Xform | Other Session -> This Session | Import tSeries only.
The pRF analysis analyses all scans within the selected data-type (e.g., Original, Averages). If you want analyze only on a subset of the scans, create a new data-type with only those scans. One way to do this is using the averageTSeries command:
VOLUME{1} = averageTSeries(VOLUME{1},[],'myNewDataTypeName');
There should be a simple routine for selecting scans and creating a new type. Any volunteers?
In the test data set that we have provided, open up the Gray view and select the Data Type 'Averages'.
You can perform the One Gaussian retinotopy analysis from the user interface you would
- Set the parameters (Analysis | Retinotopic Model | Set Parameters)
- Load the parameters (Analysis | Retinotopic Model | Load Parameters)
- View stimulus (Plot | Retinotopic Model | View Stimulus)
- Run analysis (Analysis | Retinotopic Model | Run ) or in Unix you can call (Analysis | Retinotopic Model | Run in background )
The pRF method uses the experimental stimulus as part of the estimation process. Certain classes of stimuli (e.g., rings, wedges and bars) are used in many experiments. These stimuli can be recreated from the analysis program if you specify the parameters.
You can specify these stimuli from: Analysis | Retinotopic Model | Set Stimulus Parameters. You will get a window like the one shown below. In this example data set, the parameters are already set. Just view them and click 'OK'.
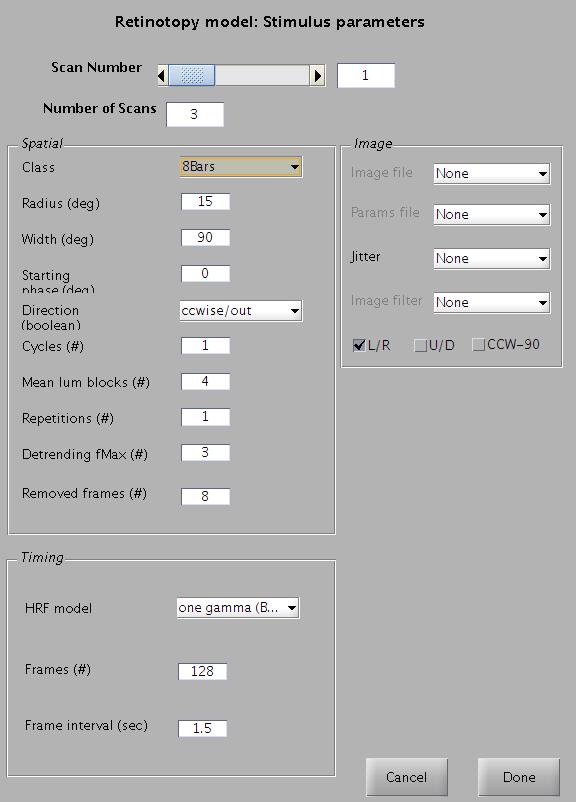
For each Scan number specify a stimulus type. Three predefined stimuli (wedge, ring or bars) are available. For each of these types you are asked to specify the stimulus size, which is the radius of the most peripheral point of the contrast signal (in deg from the fovea). You will be asked for other parameters that may be specific to that stimulus.
If you did not use one of the predefined stimuli (wedge, ring or bars), you must define the stimulus type from a stored image matrix and parameters file. To do this, choose 'StimFromScan' as the "stimulus type". You then need to choose an image file and a parameters file from the drop-down fields at the bottom of the GUI. See Creating a new stimulus.
If you used bars, the width should be set using the following formula: 360/(params.radius/params.ringDeg). Often this will be 90. The params are saved out (optionally) when running ret on the stimulus laptop. The formula above is a hack since the 8 bars code is based on originally using rings and wedges.
The stimulus window also asks you about some important non-stimulus parameters. These are essential because they depend on the stimulus and subject -
- Hemodynamic response function (HRF) model (default = one gamma Boynton-style)
- Maximum DCT frequency to be removed for detrending (default = 1)
To make a standard type of stimulus, you can create a function that makes the stimulus. Named the function make[stimName].m. For an example see makeWedge.m or make8Bars.m.
It is possible to define all of the stimulus-related (and other) parameters from the command line. Two particular important terms are the HRF and detrending parameters.
If you have specific knowledge about the subject's HRF, you can adjust either the function or function parameters.
The DCT frequency determines how the detrending is performed. The basic analysis removes low frequency DCT terms. The number you would like to remove depends on the stimulus parameters.
There are many additional parameters. An extensive set of comments and detailed description is provided in the rmDefineParameters.m file. Important parameters and examples are also summarized in rmMain.m. All the parameters have defaults in those functions. These can be changed using command line arguments.
Only the One Gaussian model can be run directly from the window interface. To run this model select
'''Analysis | Retinotopic Model | Coarse to fine minimization | Run Now '''.
This executes rmMain. This can take a long time on the whole brain.
You can run the analysis on only a region of interest. If ROIs are loaded the analysis will be performed for the current ROI only; when no ROIs exist the analysis will run on the entire dataset.
(On unix systems, you can run the analysis in the background by using Analysis | Retinotopic Model | Run in background (unix) . This function will run the code in a separate shell script - the script will not die if you quit your current matlab session or even when you logout of the machine (use kill -9 pid and typically you can find the pid using ps or top ).
You can also run the model from the command line. This is useful for scripting purposes and allows you to changing various default parameters. For example:
VOLUME{1} = rmMain(VOLUME{1},[],'coarse to fine','model',{'one gaussian','difference of gaussians'});
runs the coarse-to-fine analysis on the default model ('one gaussian') as well as a a more complex model ('difference of gaussians'). For more options see rmMain.m help rmMain and rmDefineParameters.m.
To view the results load a retinotopic model file using: File | Retinotopic Model | Select and Load Model.
You can clear this model by selecting File | Retinotopic Model | Clear.
To get basic information about the model and on when and where it was estimated try: File | Retinotopic Model | Model information.
You can visualize the parameters using File | Retinotopic Model | Load Model Parameter. You will see an interface as shown at the right.
To load the parameters from the model you specify three fields
a) Which model (if you estimated more than one), b) The parameter (e.g., eccentricity, polar angle, pRF size, variance explained) c) Which data field to store the parameter (co , amp, ph or map).
Selection (c) is obscure - sorry. The mrVista data fields have historical names that were developed for the traveling wave. We are simply storing the pRF fields into those structures. At some point we will make a more sensible arrangement.
If you don't know what the latter statement means simply leave it at the default. The default automatically stores the parameter in the most appropriate field. The default assignments are:
- co <- Variance Explained or coherence
- ph <- Polar angle
- map <- Eccentricity, pRF size, anything else
To plot the fits and the pRF for a given voxel in the current ROI use: Plot | Retinotopic Model | Receptive field and model fit | fitted timeseries.
To get the average values of a parameter in the current ROI, such as pRF size, use: Plot | Retinotopic Model | ROI stats. The average is weighted by the percent variance-explained at each voxel.
The retinotopy modeling functions generally begin with rm, as in rmMain, or rmDefineParameters.
Other important routines are the two different search strategies:
- rmGridFit
- rmSearchFit
view = rmMain([view],[roiFileName],[wSearch],[varargin]);
vw = VOLUME{1};
roiFileName = 'leftMT'; % If empty, the entire volume is fit - takes a while
searchType = 'coarse to fine';
outFileName = 'myFile'
prfModels = {'one gaussian','difference of gaussians'};
VOLUME{1} = rmMain(vw,roiFileName,searchType,'matFileName', outFileName,'model',prfModels);
The command line above illustrates the basic command line. You can adjust most of the parameters of the fit using the calling arguments. The main calling parameters you can adjust are shown by typing doc mrMain. A complete list is in the function rmDefineParameters.
removed from the data, number of Discrete Cosine functions (Transform, DCT) to remove from the data (high-pass filter, for example: 0=remove only the DC component; 3=remove up to and including three cycles per scan), and the specific hemodynamic Response Function (HRF) that you would like to use.
There are many other parameters that are set by default. The defaults can be overwritten by a command line call (see Model estimation section).
There are two ways to create a new stimulus, either by creating a new 'make' file or by using an image matrix from your scan.
It is important to note that the stimulus we make for use with retinotopic model code is not identical to a stimulus used in a scan. We use simplified stimuli for solving models. For example, there is only one image per volume acquisition (TR), and the images usually show only the stimulus aperture (e.g., a wedge), and not the carrier, or internal contrast patterns (e.g., a checkerboard). This is why we need to use special make functions.
A make* file should be named make[StimulusDescription].m and placed in the directory StimulusDefinitions. The new stimulus definition will automatically appear in the gui. Current examples include make8Bars, makeWedge, and makeRing. The make* file generates stimulus images. It takes params, a struct with stimulus parameters, as input and output, adding the field "images" to the struct. The stimulus parameters can be generated from the set parameters ui. There is one image per temporal frame, and the images are vectorized for ease of computation. The images are usually binary, with a 1 in pixel locations with non-zero contrast and 0s elsewhere. See existing make* files for further details, and rmMakeStimulus to see how the make* files are called.
Alternatively, you can make your stimulus starting with the raw images used in the scan. To do this, you need two files, an image file and a params file. The file names should contain the strings 'image' and 'params', respectively, and the files should be placed in a directory called 'Stimuli', inside the main project directory.
Both the image file and the params file can be made automatically if you use our expTools software for stimulus presentation. For example, if you run 'ret', our code for presenting retinotopic stimuli, you can check the box 'Save Stim Parameters', and enter a file name in the field 'Store image matrix' (see figure on right). This will result in two files, a parameters file and an image matrix file. The parameters file name is derived from the date and time you ran the program (e.g. 20080717T161217.mat)
If you do not use expTools to run your experiment in the scanner, or if you want to create arbitrary new stimuli, you can generate the image file and params files manually. The image file must have a variable named images, containing all the unique images shown in the scan. It is n * m * k, where n and m are the image dimensions and k is the number of images. The parameters file must have a variable named stimulus containing a field seq (stimulus.seq), and seqTiming (stimulus.seqTiming). Seq is a vector indexing the image matrix. seqTiming is a vector of the same length as seq indicating onset times of each image in seconds (zero-based, i.e. the first time point of the scan is zero). See makeStimFromScan for additional optional fields and futher details.
Once your image and params files are in the Stimuli directory, you can make your stimuli via the set parameters ui.
In the GUI, as shown on the right, select StimFromScan for Class. This will cause a number of stimulus fields to gray out; they will not be needed since the stimulus will be defined from the images themselves. The fields 'Image file', 'Params file', and 'Image filter' will be un-grayed, as these fields become necessary when defining your stimulus. The image filter is used to convert the raw images into stimulus predictors. The binary filter is most similar to the processing used in our standard make* functions, such as make8Bars, but other filters are possible.
If you have eye movement data you can alter your stimulus to compensate for eye position. To do this, put a file containing the string jitter inside the Stimuli directory. From the set parameters ui, you can then select a jitter file from the drop down menu. Eye movement jitter can be used with any kind of stimulus, whether made from a standard make* file or from your own image matrix.
The main reason why the pRF model runs in the "Gray" view is because a coarse-to-fine algorithm is implemented with two stages. In the first stage the data pRF parameter estimates are obtained from data that is smoothed across the cortical surface. In the second stage, these estimates are refined without any smoothing of the data. This approach allows for both a speed improvement as well as a more robust estimate of the pRF parameters. To be able to smooth across the cortical surface the data has to be in the "Gray" view.
From stored pRF models we create simulated data sets. These simulations have several uses. We can use this method to predict the response to novel stimuli. We can use the simulations to predict the consequences of eye movements, scotomas or other neurological defects.
This section reviews how use retinotopic model (RM) to calculate a simulated data set.
The key function for the simulation is rmCalculatePredictions. The simulation also uses rmEditStimParms, which calls a sequence of functions including rmLoadParameters and rmMakeStimulus. (These routines should probably be renamed, too).
The method of calculating a simulation from the GUI is
- Load an existing RM from the File | Retinotopy Model | Select and Load Model pull down
- From the Analysis | Retinotopic Model | Set Parameters pull down, set the properties of the stimulus for your scans
- Choose the Analysis | Retinotopic Model | Calculate pRF Predictions pull down; a new dataType will be created (default: Simulated).
When you open a data set that has a retinotopy model calculated, you can load it from File | Retinotopy Model | Select and Load Model. This will attach a structure (rm) to the VOLUME{} cell array for that window. For example,
>> VOLUME{1}.rm
ans =
retinotopyModelFile: 'Y:\users\Netta\HH080213b\Gray\Averages\retModel-20080214-221919-sFit.mat'
retinotopyModels: {[1x1 struct]}
retinotopyParams: [1x1 struct]
modelNum: 1
To simulate data, we might want to use the model and a different stimulus. For example, Jon does this when he estimates the model using retinotopy and then predicts the response to a visual brightness illusion.
THe next two steps then involve (a) creating and loading a new stimulus, and (b) combining the stimulus with the model to produce a simulated data set (prfSavePredictions).
To specify a new stimulus, you can use the function
params = rmEditStimParams(getCurView);
To load a new stimulus you can either use the current method of generating stimuli in the GUI to create a new stimulus and put it in the retinotopy model structure. The pulldown is Analysis | Retinotopic Model | Set Parameters which brings up a window where you define your new stimulus. When you are done, click OK and (a) the stimulus will be generated, and (b) the parameters will be stored in the view structure. The stimulus itself will replace the current stimulus in the rm.retinotopyParams.stim, so be careful not to over-write the existing model (this would be hard).
To be done
- We need a script to do all of this
curModel = viewGet(VOLUME{1},'rmCurrent') curModelParams = viewGet(VOLUME{1},'rmStimParams')
rmCalculatePredictions;
There is some confusing terminology
- Repetitions: If you have 6 stimulus cycles and 4 blanks then the stimulus repeats twice. SOD accounts for this when solving the model. He averages the first and second half and fits that. This saves time. But the meaning of repetitions is confusing - cycles is the number of cycles that, say, the wedge goes around. Repetitions is the number of complete repetitions of the stimulus, including both the wedge/ring and the blanks. In the convenitonal 6 cycle 4 blank experiment the number of repetitions is 2. - Put this comment in the retinotopy model solution, too.