mrAlign
rxAlign is a script that calls the mrRx tool to align inplane anatomicals to a volume anatomy.
The assumption is that you have collected images of anatomical slices that have the same prescription as your functional data. The transformation which aligns the anatomical slices to the volume anatomy can then be applied to the functional images. Therefore, before doing the alignment it is always worth verifying that your inplanes and your functional images have the same prescription. This can be done by looking at the mean maps for each run on the inplanes in mrVista.

This guide assumes that the current directory in matlab is the session directory (the one containing the mrSession.mat file). To start rxAlign, type rxAlign at the matlab command prompt, or select *Edit | Edit/View Alignment* from a mrVista window..
rxALign
Four figures will open on screen.

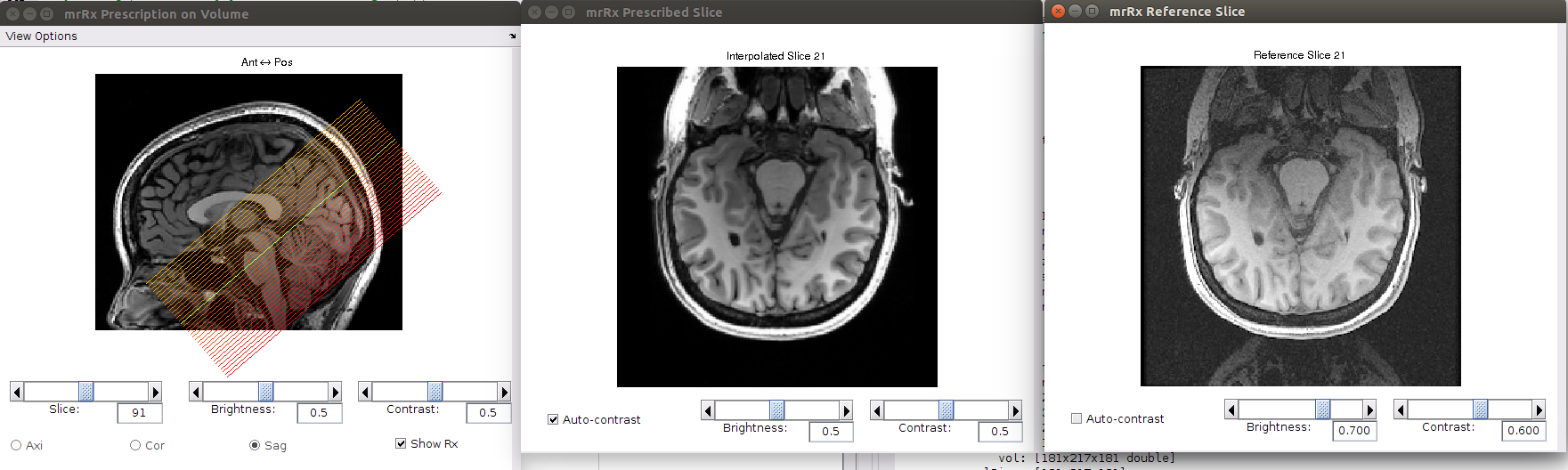
The top figure title mrRx, allows you to search for and save a transform which brings your inplanes into alignment with the volume. The bottom left figure, labeled mrRx prescription on volume, shows a slice prescription (red lines) on your volume anatomy. Adjusting the sliders in the mrRx window moves the slice prescription around. The bottom center figure (labeled mrRx Prescribed Slice) shows what an in plane given the slice prescription would look like. The inplane chosen corresponds to the yellow line in the mRx Prescription window. The bottom right window labeled mrRx reference slice, shows the corresponding inplane from your data.
In brief, you use the manual controls in the mrRx window to adjust the prescription until the mrRx Prescribed and Reference slices look as similar as possible. Then an automatic alignment algorithm is employed to fine tune the alignment. Once an acceptable alignment is achieved it is saved for use by mrVista.
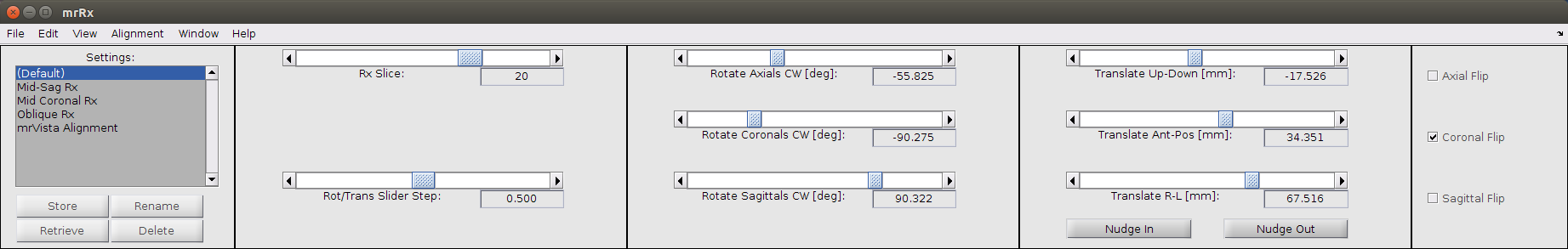
The left-most panel contains a list of default and stored alignments. The other 4 panels have controls for manually aligning the inplanes to the volumes. These are fairly easily understood by adjusting them and watching the affect on the slices in the prescription on volume window and the interpolated slice. A brief summary:
the rxSlice slider changes which slice is currently being interpolated and compared. the current slice is highlighted in yellow in the rxPrescription on volume window.
Rot/trans step adjusts how far the interpolated inplane will move when adjusting the rotation and translation sliders one step.
The rotation and translation sliders do what they say.
Checking or unchecking the boxes at right will flip the prescription in that plane (axial, sagittal, coronal).
At Lucas, there was a screen save function that would save a dicom image with the prescription on it. This image could then be put in
sessionname/Raw/Anatomy/SS/
And opened from the menubar at Window | Open Screen Save Window

Although this option may not be available to you, it is still very useful to obtain a screenshot of the functional slice prescription on the scout when you are at the scanner (usually available using print screen on the scanner keyboard). Often the most time consuming step is getting the inplanes roughly aligned with the anatomy and having an image which shows where they were on the scout can greatly speed up this process. Note that the mrRx Prescription on Volume window has three toggles at the bottom that let you see the current slice alignment in the coronal, sagittal, and axial views. This means you can get a good start on the alignment simply by moving the slices around on the volume, if you have a screenshot.
When you’re satisfied with the manual alignment, press the ‘Store’ button, on the lower left-hand side of the mrRx window, to store the current settings. You may also want to select the menu option File | Save… | mrRx settings to save out these settings to file. You can always return to those settings by highlighting them in the settings list and then clicking the restore button. You can also give them a name by highlighting them and using the rename button. It is generally a good idea to always store your manual alignment before trying one of the automatic alignment options. Sometimes when the manual alignment is insufficiently close the output of the automatic alignment will be worse than what you started with. In that case it is convenient to be able to quickly return to the previous state.
Various automatic alignment algorithms can be invoked by using the pull-down menu under Alignment. When one finishes it will create a new setting in the mrRx window at the left. If you have put some effort into the manual alignment it is worth saving that before using the algorithms as automatic alignment can sometimes end up with a worse looking alignment than the one it began with. This is more likely when the alignment between the inplanes and interpolated slices is not similar already. When this happens it saves time to be able to return previous work by highlighting the stored setting and pressing the Retrieve button.
From the menu select Alignment>>Fine>>Nestares Code. The Nestares code refers to the alignment method developed by Oscar Nestares and David Heeger, described in this paper:
The Mutual-Information alignment refers to a mutual-information based metric which uses the SPM co-registration tools (see the spm_coreg function in SPM5)
The code that performs this is rxFineMutualInf. These steps seem to produce high-quality alignments for many prescriptions. The one exception seems to be high-resolution sessions in which the prescription covers a very small part of the whole brain (e.g., part of one hemisphere). The code seems to do well for more common prescriptions using a surface coil, which only cover the posterior part of the brain (but a large amount of this cortex).
Point alignment is an option, though often not necessary. Point alignment works best when you are pretty close to a good alignment. It should always be your last step.
It is important to pick clear and reproducible points whenever you are using this feature. T-junctions in the gray matter are usually the most point-like structure in the brain.
To add points go to the pull down menu and select Edit | Points | Add Points. The the mrRx Reference Slice figure will turn yellow. Select a point on the reference slice for which you can find the corresponding point in the prescribed slice.

Select the corresponding point on the prescribed slice. Repeat the process for as many points as you want to add. Press the right mouse button to stop adding points.
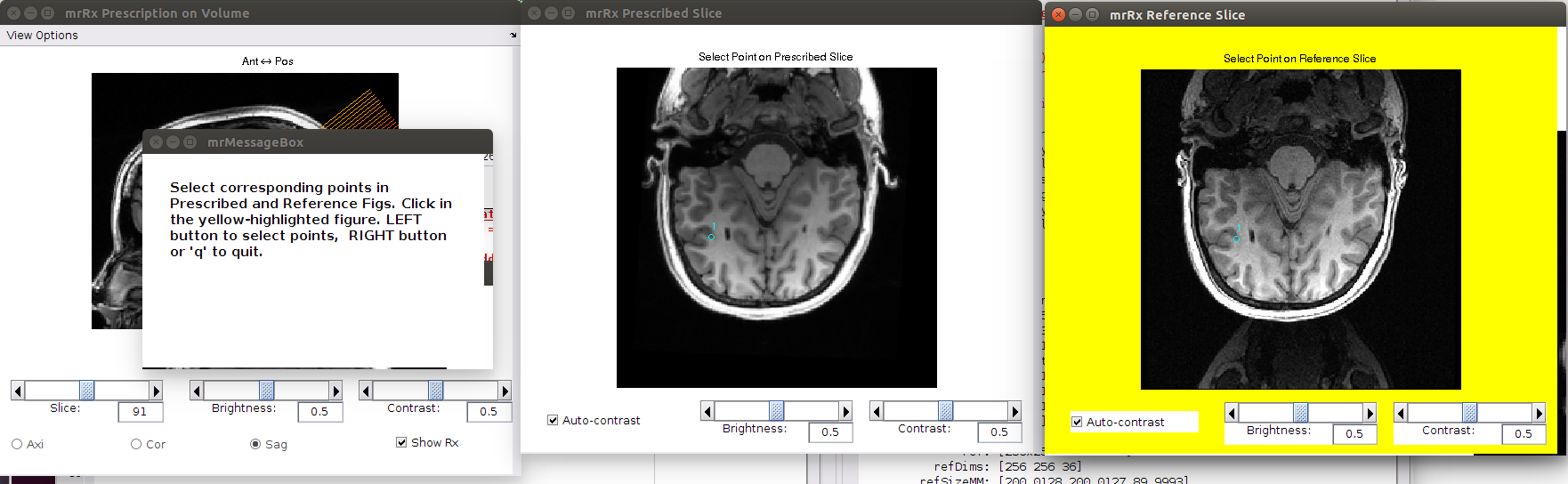
Switch to another slice, and add more corresponding points (you can do a series of even slices followed by a series of odd slices). It’s a good idea to select points in early, middle, and late slices in the inplanes, to avoid have the alignment be close for one area, but get progressively worse away from that region.
It is a good idea to store the current prescription settings, using the ‘Store’ button in the mrRx window, before computing the from-points alignment.
Select Alignment | Fine | From Selected Points. A new alignment will be created, and stored under the name Point Alignment. Feedback will be provided on the amount of error between each point and the computed ideal point. If one point has a much higher error than the other points, it was likely poorly chosen (the points on the reference and prescribed slices don’t really correspond). You can delete the points using Edit | Points | Delete Points <Ctrl-D>. A good alignment has an error of less than 1 for all points.
Choosing points can be easier and more accurate when you zoom in on a brain section.
When the same features are visible in each slice, select the menu option Window | Open Rx/Ref Comparison Window. A new figure will open up to the lower left:
Comparison methods (mrRx Compare Interp/Ref)
Overlay (red/blue): Here it is helpful to adjust the amount of red and blue contributed from each image by adjusting the Rx alpha and Ref alpha sliders until you can make out the outlines of both brains.
This figure allows you to overlay the reference and prescribed slices. Three visualizations are possible: showing the prescibed slice in red and the reference slice in blue-green, such that high overlap appears gray; showing a color-coded image of the prescribed minus the reference slice; and showing a checkerboard of alternating tiles of prescribed and reference images. These can be selected with the popup at the bottom of the figure. In addition a correlation measure, and root-mean-squared error, are shown between to the two slices. Making the slices similar entails finding a local maximum for the correlation, and a local minimum for the error.

You can toggle between two images with the toggle button at the lower right-hand corner of the comparison figure. In addition, there is a menu option on the Comparison Figure: Comparison Prefs | Compare all slices (separate GUI). A new window will open with the two volumes superimposed. You can set both volumes to a grayscale colormap using Ctrl-5; and toggle between the two quickly using Ctrl-T.
Toggling in the Rx/Ref Comparison Window is useful. The ideal would be to hit toggle, and not be able to distinguish when the toggling switches between the Rx image and the Ref image. In reality, this will never actually happen. But you can get close. For novice togglers, it might be helpful to change the number of times (line 16, nToggles), or the speed at which the toggling occurs (line 37, pause(0.15)) by editing rxToggle.m
The above methods are usually good enough. But you can also test a saved alignment in the following way (somewhat time consuming).
1. save an alignment to mrVista 2. generate a segmentation in mrVista (put link here) 3. draw an ROI on the gray view in a region you want very carefully aligned. One way would be to load the mean map and then select the region of cortex (you could pick all of it) and then restrict the ROI to gray and save. 4. open rxAlign and load the ROI. 5. see where the ROI appears on the interpolated slice.

Result of automatic alignment. These images are misaligned but it is hard to tell

The ROI here is the union of all visual field maps defined in this subject. Loading the ROI makes it obvious that the automatic alignment did not do a good job
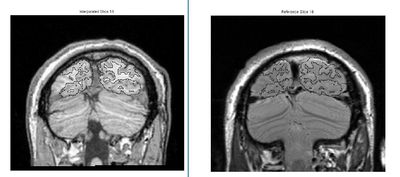
After fine manual correction, the alignment is much better. You should check other slices to make sure that the alignment is good everywhere, and not just this slice.
Check the alignment in the region or ROI you care most about Consider the alignment in the images on the right. The top image is the result of an automatic alignment. It looks OK by eye. When you load an ROI, however, you can see the error. The error is especially clear around the cortex/cerebellum boundary.
After hand-editing, the alignment is much better.
In mrRx: File | Load | mrVista ROI Lets you load a mrVista ROI which will be shown in the Rx/Ref comparison window. This should be an ROI of interest where you really need the alignment to be good. You can specifically go for the ROIs that show up as single blobs on the Inplane/Volume view, but that straddle a sulcus and break up into small pieces when shown on the mesh. A misalignment could cause a blob of activation that should be on a gyrus to shift over to a sulcus and artifically "break up" on the inflated cortical surface representation, as may be happening around the parietal-occipital fissure in the example image.
In order to see the ROI on the in the Rx/Ref comparison window, even if you toggle (usually they disappear on toggling), add the following code to rxToggle.m (written by JW)
% -------------------------------------------------------
% -- JW ADD to show ROIs
% get ROI info if it exists
rxSlice = get(rx.ui.rxSlice.sliderHandle,'Value');
rxSlice = round(rxSlice);
prefs.method = 1;
prefs.lineWidth = 1;
for R = rx.rois
prefs.color = R.color;
roiCoords = vol2rx(rx, R.volCoords, 1);
inSlice = find(round(roiCoords(3,:))==rxSlice);
end
% -------------------------------------------------------
If you have to re-save a new alignment for a subject, there are several steps you need to take:
If this person has already had ROIs made and they have been saved in the local directory, you need to make sure that you save them to the shared directory because 'everything in the Gray directory will get deleted'. You can do this moving the ROIs from the Gray/ROIs folder into the 3DAnatomy/ROIs folder.
Install Alignment: If everything you need in the Gray directory has been backed up, you can save the new alignment.
Reinstall Segmentation: From the Inplane window, Gray | Gray/White Segmentation | Install/Reinstall Segmentation You also have the option of reinstalling the segmentation with all gray nodes (otherwise, it will only create nodes in the areas where your inplane falls).
Note: Reinstalling the segmentation will also 'delete everything in the Gray directory'.
Retransform Maps<: You will need to retransform all the maps in the dataTYPE from the Inplane to the Volume again since they were a casualty of reinstalling the alignment/segmentation. From the Volume window and while you are on the GLMs dataTYPE, Xform | Inplane->Volume | All Maps in Data Type | Trilinear Interpolation'
When you are satisfied with your alignment click Store in the lower left part of the mrRx window. I generally rename this final alignment something like 'mrVistaAlign' using the Rename button. Then go to the menu and select File | Save | mrVista Alignment. This will add your alignment to the mrSession variable, with the matrix added to mrSESSION.alignment.
mrRx (for “mister prescribe”) is a Matlab tool for applying a slice prescription onto an anatomical volume. This slice prescription can be described mathematically as an affine transformation matrix. This matrix maps between coordinates in the original volume matrix, and a new coordinate space, which we call the "prescription".
Here's a rough guide to how the mapping works: suppose you have a set of coordinates in your inplane anatomy, which you keep in a matrix you call InplaneCoords. InplaneCoords is a matrix with 3 rows and several (N) columns: each column is the (row, column, slice) position of a voxel in the inplane anatomy data matrix. What you want to know is, what is the set of coordinates in my reference volume anatomy which match these inplane coordinates? We can name this desired matrix VolumeCoords. The mapping we want would be:
VolumeCoords = Xform * [InplaneCoords;]
In this case, Xform is the 4x4 affine matrix that you edit in mrRx, and the ones vector is just a vector of ones, one for each column in VolumeCoords.
One note about this: The mrSESSION.alignment matrix applies transformations in which the rotations are relative to the point (1,1,1), which is the upper left-hand corner of the first inplane slice. mrRx always rotates about the center of a volume; but again, this is accounted for when the alignment is saved.