07. Copy Number Variation detection
The Copy Number Variants (CNV) detection is performed using the tool CopywriteR published by Kuilman et al. (Genome Biol, 2015). The principle is that regions at ChIP peaks are masked and the background reads left in a user-defined bin (e.g., 10kb) are compared to the overall background signal.
Besides the CNV detection, the RPGC normalized signal will be corrected (only the targets/IP) for the presence of CNVs by dividing raw signal by the CNV scores (linear values).
| Parameter | Description |
|---|---|
| call_CNV |
True/False to indicate whether to perform the CNV detection. |
| kb_bin_resolution | Default: 20(kb). Size of the bins, in kilobase-pairs, in which evaluate the CNV scores. |
| CNA_threshold | Default: 2. Number (linear scale, absolute value) indicating the minimal copy number used to define whether detected CNV is actually a CNV. |
| CNA_plot_line_colors | Default: red. String indicating an R-supported color (see ) used for the threshold lines in the remade CNA-plot. |
| CNA_plot_point_size | Default: 0.5. Number indicating the point size (R format) to use for the remade CNA-plot. |
| CNA_plot_point_transparency | Default: 0.5. Number (between 0-1) indicating the point transparency (R format: alpha) to use in the remade CNA-plot. |
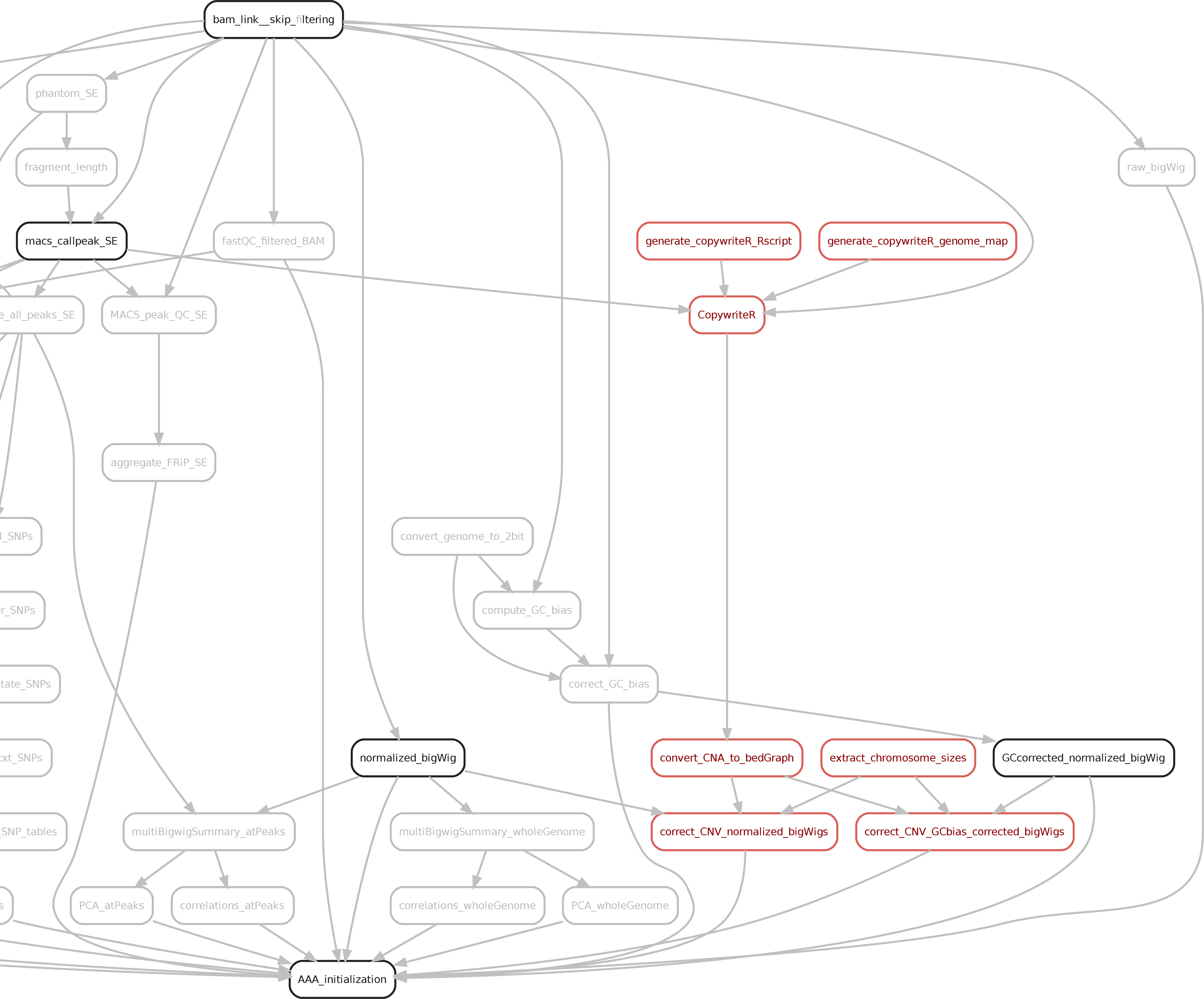
The results of the CNV detection can be found the in the folder 06b_Copy_Number_Variation. This folder will contain a subfolder for each sample including: a) a remade plot with the CNV scores for all chromosomes; b) a CNA_profiles subfolders with CNV scores plots for each chromosome, as well a bedgraph/bigwig with raw (.igv) and filtered (.bedGraph/.bw) CNV scores. Furthermore, normalized bigwig corrected for the CNV can be found the in the 03_bigWig_bamCoverage/RPGC_normalized_CNA.corrected directory.
Here an example directory tree:
output_folder
...
│
├── 03_bigWig_bamCoverage
│ ├── raw_coverage
│ │ └── ...
│ ├── RPGC_normalized
│ │ └── ...
│ ├── RPGC_normalized_CNA.corrected
│ │ └── sample_mapq20_mdup_RPGC.normalized_bs10_CNA.corrected.bw
│ ├── RRPGC_normalized_GC.corrected
│ │ └── ...
│ └── RPGC_normalized_GC.corrected_CNA.corrected
│ └── sample_mapq20_mdup_RPGC.normalized_bs10_GC.corrected_CNA.corrected.bw
...
└── 06b_Copy_Number_Variation
├── hg19_20kb #genome segmentation
│ ├── blacklist.rda
│ └── GC_mappability.rda
└── sample
├── CNA.plot_sample_all.chr_20kb.pdf
└── CNA_profiles
├── plots
│ ├── log2.sample_mapq20_mdup_sorted.bam.vs.log2.sample_mapq20_mdup_sorted
│ │ └── ... # ignore this content
│ └── log2.sample_mapq20_mdup_sorted.bam.vs
│ ├── all_chrom.pdf
| ├── chrom_1.pdf
| └── ...
├── qc
| ├── fraction.of.bin.sample_mapq20_mdup_sorted.bam.pdf
| └── read.counts.compensated.sample_mapq20_mdup_sorted.bam.png
├── sample_filtered.abs.2_linear_CNAcounts_sorted.bedGraph
├── sample_filtered.abs.2_linear_CNAcounts.bw
├── input.Rdata
├── log2_read_counts.igv
├── read_counts.txt
└── segment.Rdata
Contributors